Mexohar (Mexiletine) Monograph



Overview of Ventricular Arrhythmia (VA)
Introduction
Cardiac arrhythmias refer to conditions in which the electrical activity of the heart becomes too slow, too fast, and/or too irregular.1 Slow heart rhythms can be readily treated with electronic pacemakers, but rapid heart rhythms that culminate in atrial fibrillation (AF) or ventricular fibrillation (VF), in which the electrical activity in the atria or ventricles becomes turbulent, remains the leading causes of morbidity and mortality, including sudden cardiac death, particularly in industrialised countries.1
Mechanisms Involved
Arrhythmias can be understood as an altered function of conductive cells in the heart. Immediately after the conductive cell have depolarised, absolute refractoriness is present. This is followed by a period of relative refractoriness. By the end of the repolarisation phase, evidenced by the T wave on the electrocardiogram (ECG), refractoriness (ECK) usually has resolved. Particularly in the diseased heart, cells in the same anatomic area may repolarise at different rates.2 Arrhythmias usually occur as a result of processes that affect depolarisation, repolarisation, or conduction. These processes involve the concentration of critical cations. The three major intracellular cations are potassium, magnesium, and calcium. Changes in the concentration of any of these electrolytes may, therefore, change the risk of arrhythmia.2
There are basically three mechanisms that can cause arrhythmias:
1. Re-entry
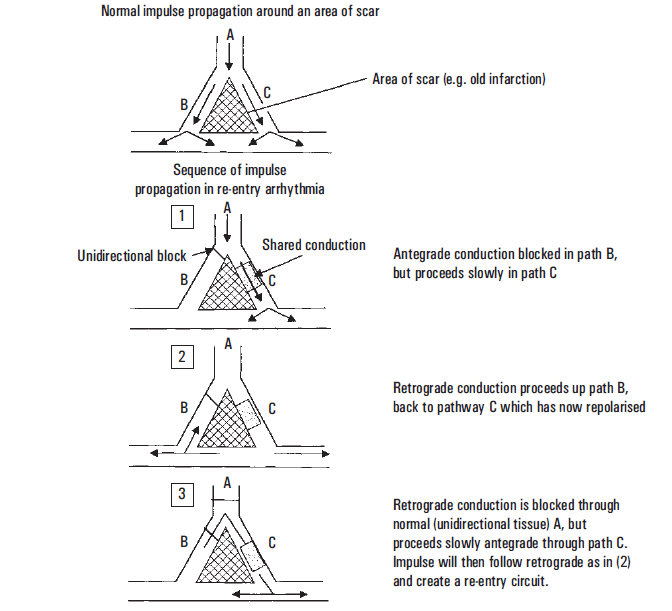
Re-entry is the most frequent mechanism causing serious VA. Re-entry requires two conditions in separate pathways. One pathway must have a unidirectional block (B), and the other pathway must have slow conduction (C). An impulse encounters an area of tissue (B) that blocks conduction in the normal (or antegrade) direction. This area (B) only conducts in a retrograde direction, that is, opposite from the normal direction. When the impulse conducted through the surrounding tissue reaches the distal portion of the unidirectional block, retrograde conduction occurs. If the slow pathway (C) is no longer refractory to depolarisation, then pathway (C) can again be depolarised. The result is a recurring circuit of depolarisation between the slowly conducting tissue (antegrade) and tissue with the unidirectional block (retrograde). Re-entry is the postulated mechanism for VT occurring in a patient with an established scar from prior infarction. In this situation, the scar and ischemia produced by the infarct create isolated areas of differential conduction that form the re-entry circuit.2
2. Automaticity
The second mechanism of VA formation is abnormal automaticity. Automaticity refers to the spontaneous depolarisation of cells due to phase 4 depolarisation. Normal automaticity occurs in pacemaker cells. Abnormal automaticity refers to spontaneous depolarisation of cells that do not normally demonstrate pacemaker function. Abnormal automaticity is seen in ischemia, disturbances in electrolytes and from various drugs such as those causing catecholamine stimulation.2
3. Triggered Activity
The third pathophysiologic mechanism for the formation of VA is triggered arrhythmia. Triggered arrhythmias, like automaticity, result from abnormal impulse formation. This tends to occur in clinical scenarios similar to automaticity. Triggered arrhythmias are caused by altered cellular depolarisation. Altered depolarisation occurs after the end of the absolute refractory period. These arrhythmias usually are associated with intracellular Ca2+ excess and can be seen during reperfusion in acute infarction or with digitalis toxicity. Fast heart rates enhance this arrhythmia and beta-blockers may help to prevent them.2
Types
Arrhythmias differ from normal heartbeats in speed or rhythm. Arrhythmias are also grouped according to the location of their occurrence, in the upper chambers of the heart, in its lower chambers, or between the chambers.3 The main types of arrhythmia are:
- Bradyarrhythmias
- Premature arrhythmias
- Supraventricular arrhythmias
- VA
These arrhythmias start in the heart’s lower chambers. They can be very dangerous and usually require medical care right away.
VF occurs if disorganised electrical signals make the ventricles quiver instead of pumping normally. Without the ventricles pumping blood to the body, sudden cardiac arrest and death can occur within few minutes.3
Definition of Ventricular Tachycardia (VT)
VT is defined as fast, regular beating of the ventricles that may last for only a few seconds or for much longer. A few beats of VT often do not cause problems. However, episodes that last for more than a few seconds can be dangerous. VT can turn into other more serious arrhythmias, such as VF, or v-fib. Torsades de pointes is a type of arrhythmia that causes a unique pattern on an EKG and often leads to vfib.3
Epidemiology of VT
Over the past few decades there has been a steady decline in the rates of fatal VF and VT. VT was previously estimated to account for up to three quarters of out-of-hospital cardiac arrests. The Framingham Study demonstrated a 49% decrease in age-adjusted risk from 1950–1969 to 1990–1999. From PANARM-HF registry, VT/VF contributed to 4.5% of patients with heart failure and VA.4
Age-related Demographics
VT is unusual in children but may occur in the post-operative cardiac setting or in patients with associated congenital heart disease. Tachydysrhythmias in children is more commonly due to paroxysmal supraventricular tachycardia’s (PSVTs). The incidence of ischemic VT increases with age, regardless of sex, as the prevalence of coronary artery disease (CAD) increases. VT rates peak in the middle decades of life, in concert with the incidence of structural heart disease. Idiopathic VT can be observed at any age.5
Sex-related Demographics
VT is observed more frequently in men because ischemic heart disease is more prevalent in men. Among patients with CAD in the Framingham Heart Study, male deaths were more common than female deaths (46% vs 34%, respectively). It seems certain that as CAD becomes more common in women, the incidence of VT in women will increase.
Females with acquired or congenital long QT syndromes are at greater risk for sudden death. The opposite is true for arrhythmogenic right ventricular cardiomyopathy (a two-fold male predominance) and Brugada syndrome (an approximately eight-fold male predominance).5
Pathogenesis of VT
Premature ventricular complexes (PVCs) are often the events that lead to VT. These premature beats may occur as a result of either a change in automaticity or a conduction defect.
As a premature ventricular beat often propagates by travelling through ventricular muscle, conduction is delayed and the QRS complex on the electrocardiogram is wide. The pattern may also be one of bigeminy, a premature beat for every normal beat, or trigeminy, wherein every third beat is a premature beat. As the premature beat originates in the ventricle, it is not associated with a P wave preceding it.3
Sustained monomorphic VT sometimes appears as a narrow QRS complex on the electrocardiogram if the sinus node generates a wavefront that results in depolarisation of the ventricles prior to the effect of the premature ventricular beat, essentially cancelling the spread of the premature beat. This is called a capture beat. Sometimes, a combination of a narrow QRS complex and a wide QRS complex occurs. This hybrid QRS complex is called a fusion beat.3
Polymorphic VT has a unique pattern on the ECG and is considered with respect to torsade’s de pointes. Genetic predispositions are a risk factor for VT. As ion channel activity is the basis for the electrophysiological properties of the heart, inherited channelopathies play a role in arrhythmia susceptibility. Displayed below is an action potential in the myocardium with contributions of ion currents to its various phases.3
The relative magnitudes of the various ionic fluxes, as they apply to the cell, are shown by the size of the arrows above the action potential; ↑, a depolarising current; ↓, a repolarising current. Predominate channel subtype activities for Ca++ and K+ channels are shown above the respective arrows (T = transient calcium, L = long–lasting calcium, Kr = rapid potassium, Ks = slow potassium, Kur = ultra-rapid potassium, Kir = inwardly rectifying potassium).3
An increase in the conductance of the sodium channel can lead to long QT intervals on the electrocardiogram and torsade’s (polymorphic VT). A decrease in sodium channel opening can lead to the Brugada syndrome and greater risk of VT. Polymorphic VT is also associated with a mutation of the ryanodine receptor Ca++ channel, co-localised near T tubules, where L-type Ca++ channels also contribute to an increase in Ca++ current and thereby contribute to a risk of VT. A decrease in the function of Ks and Kr potassium channels can also predispose to long QT-associated arrhythmias. These channel mutations may be present but not manifest themselves unless there is a precipitating cause, such as a drug that prolongs the QT interval, an electrolyte imbalance, or high catecholamine levels.3
Etiology of VT
Causes of VT include the following:5
- Ischemic heart disease (most common).
- Structural heart disease with disruption of normal conduction patterns (e.g., non-ischemic cardiomyopathy, arrhythmogenic right ventricular cardiomyopathy [ARVD] or cardiomyopathy, hypertrophic cardiomyopathy [HCM]).
- Congenital structural cardiac disorders (e.g., tetralogy of Fallot) and associated surgical scar.
- Acquired channelopathies, most commonly from drugs that prolong the QT interval (e.g., class IA and class III antiarrhythmics, phenothiazines, methadone, many others); drugs that slow myocardial conduction (e.g., flecainide, propafenone, halothane) may also promote re-entrant VT.
- Inherited channelopathies (e.g., long QT syndrome, short QT syndrome, Brugada, and catecholaminergic polymorphic VT).
- Electrolyte imbalances (e.g., hypokalaemia, hypocalcaemia, hypomagnesemia).
- Sympathomimetic agents, including intravenous (IV) inotropes and illicit drugs such as methamphetamine or cocaine.
- Digitalis toxicity, which can lead to biventricular tachycardia.
- Systemic diseases causing infiltrative cardiomyopathy or scar (e.g., sarcoidosis, amyloidosis, systemic lupus erythematosus, hemochromatosis, rheumatoid arthritis).
- Hypokalaemia is an important arrhythmia trigger, followed by hypomagnesemia.
- Hyperkalaemia may also predispose to VT and VF, particularly in patients with structural heart disease. Other triggers include sleep apnoea and AF, which can increase VT risk in patients with pre-existing structural heart disease.
- QT prolongation, which may be acquired or inherited, can lead to VT. Acquired QT prolongation is observed with certain potassium channel–blocking medications. Most of the causative drugs block the delayed rectifier cardiac potassium current, IKr. These agents include class IA and class III antiarrhythmics, azithromycin, and many others. Congenital long QT syndrome is a group of genetic disorders involving abnormal cardiac ion channels (most commonly, potassium channels responsible for ventricular repolarisation).
- In both acquired and congenital long QT syndromes, prolonged repolarisation predisposes to torsade de pointes, a re-entrant rhythm with a constantly varying circuit. Other inherited ion channel abnormalities may cause idiopathic VF and familial polymorphic VT in the absence of QT prolongation.
Among patients younger than 35 years, the most common cardiac causes of sudden death, and presumably of VT, include the following:
- HCM
- Right ventricular cardiomyopathy (ARVD)
- Myocarditis
- Long QT syndrome
- Congenital coronary artery abnormalities
- Inherited long QT syndrome
- Catecholaminergic polymorphic VT
- Dilated cardiomyopathy
- Arrhythmogenic right ventricular dysplasia
- Familial VT
Symptoms and Presentation of VT
Brief episodes of VT may not cause any symptoms in some people. Others may experience:5
- Dizziness
- Shortness of breath
- Light-headedness
- Feeling as if your heart is racing (palpitations)
- Chest pain (angina)
- Seizures
Sustained or more serious episodes of VT may cause:5
- Loss of consciousness or fainting
- Cardiac arrest (sudden death)
Diagnosis of VT
Screening
Screening of first-degree relatives should be contemplated when a patient is identified as having any of the following:
- Long QT syndrome
- Short QT syndrome
- Hypertrophic or dilated cardiomyopathy
- Right ventricular dysplasia
Family screening typically involves the following:
- History and physical examination
- ECG
- Echocardiography
- Treadmill testing
In some patients with spontaneous polymorphic VT, genetic studies may be helpful for family screening or for clarifying a diagnosis. Spontaneous polymorphic VT may be related to genetic mutations affecting ion channels, such as those that occur in long QT syndrome, Brugada syndrome, and catecholaminergic polymorphic VT. Finally, some patients are predisposed to drug-induced VA by otherwise subclinical genetic ion channel defects.
Chest radiography is indicated if symptoms suggest the possibility of heart failure or other cardiopulmonary pathology as a contributing factor. Cardiac computed tomography (CT) scanning and cardiac magnetic resonance imaging (cMRI) are evolving quickly but have not yet supplanted echocardiography and nuclear imaging for quantification of ventricular function. cMRI can be especially helpful in the evaluation of uncommon myocardial infiltrative diseases, such as sarcoidosis.5
Laboratory Studies
Assess electrolyte levels of all patients with VT, including serum potassium, magnesium, calcium, and phosphate levels. Ionised calcium levels are preferred to total serum calcium levels. Hypokalaemia is a common VT trigger and is commonly seen in patients taking diuretics. Hypokalaemia, hypomagnesemia, and hypocalcaemia may predispose patients to either monomorphic VT or torsade de pointes.5
In accordance with the clinical history, measure serum levels of therapeutic drugs (e.g., digoxin, tricyclic antidepressants). Toxicology screens (e.g., for methamphetamine, methadone, cocaine) may be helpful in cases related to recreational or therapeutic drug use.
Evaluate for myocardial ischemia or infarction with serum cardiac troponin I or T levels or other cardiac markers if symptoms or clinical signs of ischemia are present. Persistently elevated cardiac enzyme levels may also be an indication of ongoing myocarditis.5
Electrocardiography
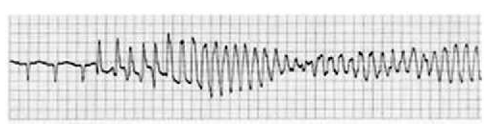
Polymorphic VT
When the QRS complex varies from beat to beat, the rhythm is described as polymorphic VT and suggests a variable electrical activation sequence. The most notorious, and probably the most common form of polymorphic VT is torsade de pointes, a French term meaning “twisting of the points” and refers to the unusual shifting-axis QRS complexes that appear as if the heart is rotating upon an axis.5
The typical initiation of torsade de pointes occurs with a “long-short” sequence—that is, a longer RR interval resulting in further prolongation of the QT interval, followed by an early depolarisation occurring at a time of heterogeneous repolarisation.5
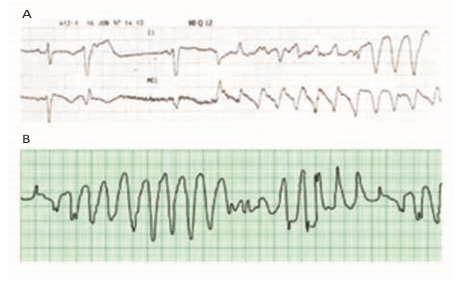
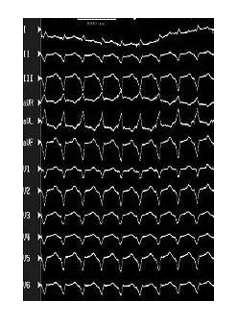
Monomorphic VT
When the ventricular activation sequence is constant, the ECG pattern remains the same, and the rhythm is called monomorphic VT. Monomorphic VT is most commonly seen in patients with underlying structural heart disease. There is typically a zone of slow conduction, most commonly the result of scarring or fibrillar disarray. Causes include prior infarct, any primary cardiomyopathy, and surgical scar, hypertrophy, and muscle degeneration.5
Monomorphic VT is occasionally observed in patients with structurally normal hearts (idiopathic VT). These VTs are often exercise dependent, and their clinical behaviour may be more consistent with triggered activity or abnormal automaticity.5
Monomorphic VTs are typically named for their site of origin. The following are the most commonly involved sites:
- Right ventricular outflow tract
- Left ventricular outflow tract
- Left ventricular septum
- Aortic root
The QRS morphology during VT can be used to predict the exit site from the zone of slow conduction or the site of origin, regardless of the underlying substrate. The earliest activation is closest to the leads with QS complexes during tachycardia.5
Monomorphic VTs have classically been considered benign. Rarely, however, they may result in sudden death, despite the presence of a structurally normal heart.5
Echocardiography
Echocardiography is used for patients at high risk for serious VA or sudden cardiac death. In particular, echocardiography can provide an estimate of left ventricular (LV) systolic function, and the presence or absence of associated LV wall motion abnormalities commonly indicative of a prior scar. Echocardiography may also show findings suggestive of a myocardial infiltrative process. Imaging of the right ventricle (RV) may be more limited, and other imaging techniques may be required to obtain accurate and global views of RV function.5 The high-risk group consists of patients with any of the following:5
- Dilated, hypertrophic, or RV cardiomyopathy
- A history of acute myocardial infarction
- Inherited disorders associated with sudden cardiac death
Cardiac Imaging Studies
Cardiac CT scanning and cMRI are evolving quickly but have not yet supplanted echocardiographic and nuclear imaging for quantification of ventricular function. cMRI can be especially helpful in the evaluation of uncommon myocardial infiltrative diseases, such as sarcoidosis.5
The use of late gadolinium enhancement (LGE) and extracellular volume (ECV) cMRI appear to have the potential to predict the estimated 5-year risk of sudden death and syncope or non-sustained VT in patients with HCM.5 In a study of 73 German patients with HCM and 16 control subjects, investigators found that not only was global ECV was the best predictor of an increased risk of sudden death but that when used in conjunction with the sudden cardiac risk score, the diagnostic accuracy to identify HCM patients with syncope or non-sustained VT was significantly improved. These findings may have implications for improved patient selection of HCM patients for implantable cardioverter-defibrillator (ICD) implantation.5
Although cMRI is often used for the evaluation of arrhythmogenic right ventricular dysplasia, the diagnostic yield of this test has yet to be clearly defined. Right ventricular angiography may still be the criterion standard imaging study for this disorder.
MRI, cardiac CT scanning, or radionuclide angiography can be useful in patients with VA when echocardiography fails to provide an accurate evaluation of left or right ventricular function. These studies may also be useful for assessment of structural changes in the heart.5
Pacemaker and other Cardiac Devices
The presence of a dual-chamber pacemaker or ICD can occasionally simplify the diagnosis. Most contemporary devices are capable of recording and logging tachyarrhythmias for subsequent analysis during an interrogation of the implanted device, as well as providing real-time telemetry of intracardiac signals.5
Analysis may reveal the disease process underlying the VT. However, the episode may prove to have been triggered by the device itself. Possibilities include the following:
- Tracking of an atrial tachyarrhythmia in a dual-mode, dual-pacing, dual-sensing (DDD) device or an atrial-triggered, ventricular-inhibited (VDD) device
- Endless loop tachycardia
- Inappropriate rate-responsive pacing due to sensor problems or incorrect sensor programming
- Overt pacemaker failure (runaway pacer)
The most common problem involves the patient whose device is tracking AF or atrial flutter. In the absence of a mode-switching algorithm, a DDD or VDD pacer responds by pacing the ventricle at the programmed upper rate limit of the device. Application of a magnet to the pacer generator may terminate endless loop tachycardia or drop the paced rate enough to allow diagnosis of the underlying atrial tachyarrhythmia.5
Other Diagnostic Considerations
VF is a disorganised, rapid ventricular rhythm that varies in interval and waveform. It may be difficult to distinguish rapid from polymorphic VT. Sudden death accounts for approximately half of all deaths from cardiovascular disease and is generally caused by VT and VF. 6
Accelerated idioventricular rhythm is defined as an enhanced ectopic ventricular rhythm with at least three consecutive ventricular beats that is faster than the normal intrinsic ventricular escape rhythm (≤40 beats/min) but slower than VT. However, there is a potential definitional overlap with accelerated idioventricular rhythm and an automatic VT of 100–120 beats/min.
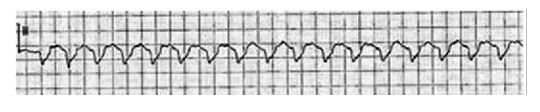
Pacemaker-generated Tachycardia.6
Permanent pacemakers occasionally generate rapid rhythms. The most common cause is tracking of atrial tachyarrhythmias, such as atrial flutter or AF. The pacemaker typically paces around the programmed maximum tracking limit, which is often set at 120–40 beats/min in older patients (see the image below). If a pacemaker programmer is not available, a magnet placed over the pacer generator deactivates atrial sensing temporarily and allows for the diagnosis of the atrial arrhythmia.6
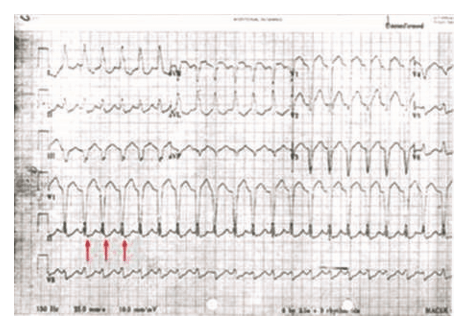
Supraventricular Tachycardia (SVT)
Wide-complex conduction during SVT can mimic VT. The two most common forms are atrioventricular re-entrant tachycardia (AVRT) and atrioventricular nodal re-entrant tachycardia (AVNRT) with aberrant conduction.6
AVRT can be either orthodromic or antidromic, depending on the direction of conduction through the AV node. All antidromic AVRTs cause wide-complex tachycardia as a result of ventricular activation outside of the His-Purkinje system, and some orthodromic AVRTs conduct with wide QRS complexes as a result of functional or pre-existing bundle-branch block, bystander accessory pathway conduction, or intraventricular conduction delay.6
- Aberrantly conducted SVT circuits can mimic VT, but careful analysis of the ECG can allow discrimination of VT from aberrant SVT in most cases.
ECG is the criterion standard for the diagnosis of VT. In a patient who is hemodynamically unstable or unconscious, however, the diagnosis of VT is made from the physical findings and ECG rhythm strip only.5
Advanced cardiac life support (ACLS) protocols should be quickly followed. Laboratory tests should be deferred until electrical cardioversion has restored sinus rhythm and the patient is stabilised. If the patient is hemodynamically stable at presentation, a 12-lead ECG and electrolyte levels may be obtained before attempting conversion with medications or direct current (DC) cardioversion. Note that if electrolyte levels are not obtained in an acute evaluation of VT post conversion, the hyperadrenergic state or hemodynamic compromise often associated with VT may affect the subsequently obtained electrolyte laboratory values.5
The ECG should be repeated once sinus rhythm has been restored, or when prior VT is suspected, as in a patient who experienced syncope. The ECG may also provide clues for differentiating among potential arrhythmia mechanisms or causes of VT, such as the following:
- Acute or chronic infarction
- Ischemia
- Myocardial scar
- Ventricular pre-excitation
- Hypertrophy
- Conduction disease
- QT prolongation
- Other precordial repolarisation abnormalities (e.g., Brugada syndrome, ARVD)
Treatment of VT
Pharmacological Cardioversion
Sustained VT may lead to hemodynamic collapse. Consequently, these patients require urgent conversion to sinus rhythm. The strategy for conversion depends on whether the patient is hemodynamically stable or unstable.
Unstable patients have signs or symptoms of insufficient oxygen delivery to vital organs as a result of the tachycardia. Such manifestations may include the following:
- Dyspnoea
- Hypotension
- Altered level of consciousness
In the workup, this situation must be differentiated from clinical manifestations of an underlying medical condition that is causing secondary tachycardia.
Unstable patients with monomorphic VT should be immediately treated with synchronised DC cardioversion, usually at a starting energy dose of 100 J (monophasic; comparable biphasic recommendations are not currently available). Unstable polymorphic VT is treated with immediate defibrillation. The defibrillator may have difficulty recognising the varying QRS complexes; therefore, synchronisation of shocks may not occur.7
Stable patients have adequate vital end-organ perfusion and thus do not experience signs or symptoms of hemodynamic compromise. Treatment depends on whether the VT is monomorphic or polymorphic and whether LV function is normal or impaired (e.g., reduced LV ejection fraction [LVEF], heart failure).7
In stable patients with monomorphic VT and normal LV function, restoration of sinus rhythm is typically achieved with IV procainamide, amiodarone, or sotalol. Lidocaine may also be used, but this agent may have common and limiting side-effects and, consequently, increase the overall mortality risk. A 12-lead ECG is obtained before conversion.7
If LV function is impaired, amiodarone (or lidocaine) is preferred to procainamide for pharmacologic conversion because of the latter drug’s potential in exacerbating heart failure. However, mounting evidence indicates that amiodarone should not be the first-line antiarrhythmic for stable VT, because its effects on myocardial conduction and refractoriness are gradual in onset. If medical therapy is unsuccessful, synchronised cardioversion (50–200 J monophasic) following sedation is appropriate.7
Polymorphic VT in stable patients typically terminates on its own. However, it tends to recur. After sinus rhythm returns, the ECG should be analysed to determine whether the QT interval is normal or prolonged. Polymorphic VT in patients with a normal QT interval is treated in the same manner as monomorphic VT.7
If the patient has runs of polymorphic VT punctuated by sinus rhythm with QT prolongation, treatment is with magnesium sulphate, isoproterenol, pacing, or a combination thereof. Administration of phenytoin and lidocaine may also help by shortening the QT interval in this setting, but procainamide and amiodarone are contraindicated because of their QT-prolonging effects. Magnesium is unlikely to be effective in patients with a normal QT interval.
In patients with electrolyte imbalances (e.g., hypokalaemia or hypomagnesemia from diuretic use), correction of the abnormality may be necessary for successful cardioversion. In patients with severe digitalis toxicity (e.g., with sustained VA, advanced AV block, or asystole), treatment with anti-digitalis antibody may be indicated. 7
After conversion of VT, the clinical emphasis shifts to determining the severity of heart disease, assessing the prognosis, and formulating the best long-term management plan. Options, depending on the severity of symptoms and degree of structural heart disease, include the following7:
- Antiarrhythmic medications: Effective in reducing the arrhythmia burden but have no demonstrated mortality benefit; however, results from the Amiodarone, Lidocaine, or Placebo (ALPS) study indicate poor but not invariably fatal outcomes from the use of amiodarone or lidocaine for non-shockable (asystole/pulseless electric activity)-turned-shockable (VF/VT) out-of-hospital cardiac arrest during resuscitation
- ICD: Aids in the acute termination of VA and provides information on the long-term management of patients with VT.
- Catheter ablation: Effective, but recurrent VT is not uncommon.
- Combinations of these therapies are often used when structural heart disease is present.
Antiarrhythmic drugs have traditionally been the mainstays of treatment for clinically stable patients with VT. However, some patients experience unacceptable side-effects or frequent recurrence of VT with drug therapy. As a result, cardiologists are increasingly making use of devices and procedures designed to abort VT or to remove the dysrhythmogenic foci in the heart. In patients with idiopathic VT (associated with structurally normal hearts), medications are often avoided entirely through the use of curative catheter-based ablation.7
Congenital long QT syndrome and catecholamine polymorphic VT have been linked to sudden cardiac death. Patients with these disorders are managed with a combination of genetic typing, beta blockers, lifestyle modification and, in selected cases, ICD placement.7
Initial Supportive Management
Rapid transport to an emergency department (ED) is essential. Emergency medical service (EMS) personnel may be called upon to provide cardioversion/defibrillation in the field if they have sufficient training and if appropriate protocols exist.
EMS personnel must pay adequate attention to the primary survey and address airway, breathing, and circulation as necessary. Beyond those steps, vascular access, supplemental oxygen, and electrocardiographic rhythm strip monitoring are all-important, but they should not delay rapid transport to the ED for definitive care.8
Long-term Treatment
Patients with monomorphic VT who have structurally normal hearts are at low risk of sudden death. Consequently, ICDs are rarely necessary in this setting; these patients are almost always managed with medications or ablation.8
Amiodarone is a complex antiarrhythmic drug that deserves special mention. It is generally listed as a class III agent but has measurable class I, II, and IV effects. Unlike class I antiarrhythmics, amiodarone appears to be safe in patients with left ventricular dysfunction.
Amiodarone, when used in combination with beta blockers, can be useful for patients with left ventricular dysfunction due to previous myocardial infarction and symptoms due to VT that do not respond to beta blockers.8
In the Electrophysiologic Study versus Electrocardiographic Monitoring (ESVEM) trial, which compared long-term treatment with seven antiarrhythmic drugs (not including amiodarone) in patients with VT, the risks of adverse drug effects, arrhythmia recurrence, or death from any cause were lowest with sotalol. The other antiarrhythmic drugs studied in the ESVEM trial were imipramine, mexiletine, pirmenol, procainamide, propafenone and quinidine.8
In patients with heart failure, the best-proven —albeit nonspecific— antiarrhythmic drug strategies include the use of the following:
- The beta receptor blocking drugs carvedilol, metoprolol, and bisoprolol
- Angiotensin-converting enzyme (ACE) inhibitors
- Aldosterone antagonists
Statin therapy is advantageous in patients with coronary heart disease, to reduce the risk of vascular accidents, VA (possibly), and sudden cardiac death.
Although idiopathic VTs often respond to verapamil, this agent may cause hemodynamic collapse and death when administered to treat VT in patients with left ventricular dysfunction. Therefore, verapamil (or any other calcium-channel blockers) is contraindicated in any patient with wide-complex tachycardia of uncertain aetiology.8
Catheter Ablation
Radiofrequency ablation (RFA) via endocardial or epicardial catheter placement can be used to treat VT in patients with left ventricular dysfunction from previous myocardial infarction, cardiomyopathy, bundlebranch re-entry, and various forms of idiopathic VT (see the image below). RFA is often used in conjunction with ICD therapy in the presence of recurrent VT episodes to reduce the frequency of required ICD therapies. For patients with structural heart disease, it is currently uncertain whether VT ablation obviates other therapies, such as placement of an ICD. 8
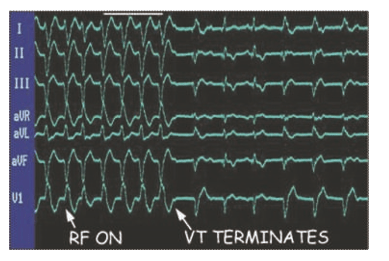
Curative ablation of VT. The patient had VT in the setting of ischemic cardiomyopathy. VT was induced in an electrophysiology laboratory, and an ablation catheter was placed at the critical zone of slow conduction within the VT circuit. Radiofrequency (RF) energy was applied to tissue through the catheter tip, and VT was terminated when the critical conducting tissue was destroyed.8
Catheter ablation is used early in patients with idiopathic monomorphic VT (i.e., VT in a structurally normal heart arising from a focal source) that is resistant to drug therapy, as well as in those who are drug-intolerant or do not wish to have long-term drug therapy. In these patients, ablation is used to treat symptoms rather than to reduce the risk of sudden death. In patients with structurally normal hearts, catheter ablation can eliminate symptomatic VT arising from the right or left ventricle.8
Catheter ablation may also be used in patients with cardiomyopathy. The goal in these cases is to reduce the arrhythmia burden and thereby minimise the number of ICD shocks.
Ablation is also used in patients with bundle-branch re-entrant VT. Most ischemic re-entrant VT requires a slow conduction zone, which is usually located along the border of a scarred zone of myocardium. The small physical size of the slow conduction zone makes it an ideal target for focal ablation procedures. Cell disruption can be achieved by using RFA or cryoablation via transvenous catheters during closed-chest procedures.8
Because patients with ischemic VT often have multiple re-entrant circuits, ablation is typically used as an adjunct to ICD therapy. If VT arises from an automatic focus, the focus can be targeted for ablation.
In patients with structurally normal hearts, the most common form of VT arises from the right ventricular outflow tract (RVOT). The typical outflow tract ectopic beat shows a positive QRS axis in the inferior leads. Abnormal or triggered automaticity is the most likely mechanism, and focal ablation is curative in these patients. Ablation cure rates typically exceed 95% if the arrhythmia can be induced in the electrophysiology laboratory. The difficulty of outflow tract ablation may be predicted by ECG morphology.8
Re-entrant tachycardia may arise from the RVOT in patients with right ventricular dysplasia or repaired tetralogy of Fallot. These circuits are usually amenable to catheter ablation.
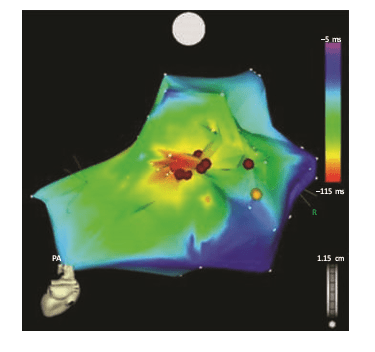
The earliest activation is recorded in red, and late activation as blue to magenta. Fragmented lowamplitude diastolic local electrograms were recorded adjacent to the earliest (red) breakout area, and local ablation in this scarred zone (red dots) resulted in termination and noninducibility of this previously incessant arrhythmia.8
Implantable Cardioverter-Defibrillator Placement
The advent of the ICD has changed the face of VA management. Like pacemakers, these devices can be implanted transvenously in a brief, low-risk procedure. Once implanted, the ICD can detect ventricular tachyarrhythmias and terminate them with defibrillation shocks or anti-tachycardia pacing algorithms.8
ICD therapy is used to augment medical management for the following individuals:
- Most patients with hemodynamically unstable VT
- Most patients with prior MI and hemodynamically stable sustained VT
- Most cardiomyopathy patients with unexplained syncope (an arrhythmia is presumed)
- Most patients with genetic sudden death syndromes when unexplained syncope is noted
In patients with prior VT or VF, the Antiarrhythmics Versus Implantable Defibrillators (AVID) study, the Canadian Implantable Defibrillator Study (CIDS), and the Cardiac Arrest Study, Hamburg (CASH), demonstrated better survival with ICD therapy than with antiarrhythmic therapy with amiodarone or sotalol. The survival difference was statistically significant in AVID, of borderline significance in CIDS, and insignificant in CASH. A meta-analysis of the three trials found a 28% reduction in relative risk of death.8
Patients with nonischemic dilated cardiomyopathy and considerable left ventricular dysfunction, or arrhythmogenic right ventricular cardiomyopathy, who have sustained VT or VF should have ICD placement. These patients should also be receiving optimal long-term medical therapy and may reasonably be expected to survive with good functional status for longer than 1 year. 8
ICDs are not used for the following individuals:
- Patients with VT or VF occurring during an acute ST-segment elevation MI (STEMI)
- Patients with reversible, drug-induced VT
- Patients with poor expected survival as a consequence of comorbid conditions
Because ICDs treat, rather than prevent, ventricular arrhythmias, as many as 50% of ICD recipients require therapy with antiarrhythmic drugs to reduce the potential for ICD shocks. Catheter ablation may be used in patients with an ICD who are receiving multiple shocks because of sustained VT that is not manageable by changing drug therapy or who do not wish to undergo long-term drug therapy.8
Nonpharmacological Cardioversion
Patients with ischemic VT may benefit from low-cholesterol diets, low-salt diets, or both. Patients with idiopathic VT may notice a reduction in symptoms when stimulants (e.g., caffeine) are avoided. Fish oil supplementation does not reduce the risk of VT or VF in patients with ICD and a recent sustained ventricular arrhythmia.9
VT may be precipitated by increased sympathetic tone during strenuous physical exertion. A goal of successful VT management is to allow the patient to return to an active lifestyle through medications, ICD implantation, ablation therapy, or some combination thereof.
Smoking should be strongly discouraged in all patients who have, or who are thought to have, VA, aborted sudden cardiac death (SCD), or both. Cigarette smoking is an independent risk factor for SCD, typically from arrhythmia and regardless of underlying coronary heart disease; smoking cessation significantly reduces the risk of SCD.9
Mexiletine in Management of VT
Mechanism of Action
Mexiletine is a sodium channel blocker and further classified as a Class 1B antiarrhythmic in the Vaughan-Williams classification scheme of antiarrhythmic drugs. Mexiletine is an oral medication that blocks sodium channels in cardiac myocytes and nerve cells. Relative to cardiac myocytes, Mexiletine effects phase 0 of the cardiac myocyte action potential, inhibiting the inward sodium current, and its mechanism of action can be generalised across all Class 1B antiarrhythmics. The uniqueness of this subclass of sodium channel blockers is characterised by the type of sodium channels they best bind to and their effect on the cardiac action potential with relative changes on EKG. To further understand the properties of the Class 1B drugs, it is important to review the cardiac myocyte action potential. The action potential is broken into five separate phases, phases 0 to 4, where the sodium channels are responsible for the depolarisation from the negative, approximately -85 to -90 mV, resting membrane potential to a positive depolarised state, termed phase 0 of the cardiac action potential. The class 1B antiarrhythmics bind best to sodium channels in the depolarised state. This correlates with the primary use of class 1B antiarrhythmics and their value in ischemic arrhythmias. Ischemia causes damage to the cardiac myocytes, rendering them unable to maintain their negative resting membrane potential, producing an increased number of cardiac myocytes in the depolarised state. Moreover, the class 1B drugs have the least effect on the cardiac action potential compared to the other class 1 drugs, likely secondary to the specificity of their binding.10
Furthermore, most Class 1 antiarrhythmics exhibit a property called "use dependence". Use dependence is named in relation to the state of the channel that is being affected. Class 1 antiarrhythmic drugs block sodium channels best when these channels are in use, or specifically when the sodium channel is in an open or inactive state. Cardiac myocyte sodium channels are in these states more often at faster heart rates, allowing more drug to bind at these higher rates. Mexiletine exhibits use dependence by rapidly unbinding when the myocytes re-polarise and no longer remain in the depolarised state, or when the sodium channels are in a resting state (not in use). During faster heart rates, the duration between depolarised states is reduced, decreasing the time to allow the drug to unbind and increasing the time sodium channels spend in an open or inactive state, which leads to more drug bound to sodium channels, and thus having a greater effect. At slower heart rates, there is more time between subsequent cardiac myocyte action potentials, allowing more of the drug to unbind as the sodium channels spend more time in the resting state. Simply put, use dependence means more binding at faster heart rates, and its clinical relevance is that tachycardia can be dangerous as there is an increased risk of adverse effects and toxicity at these faster rates.
Relative to nerve cells, blocking sodium channels disrupts the resting membrane potential, causing an inhibition of propagation of electrical impulses.10
Electrophysiology
Mexiletine is a Class 1B antiarrhythmic compound with electrophysiologic properties in man similar to lidocaine, but dissimilar from quinidine, procainamide, and disopyramide.
In patients with normal conduction systems, Mexiletine HCl has a minimal effect on cardiac impulse generation and propagation. In clinical trials, no development of second-degree or third-degree AV block was observed. Mexiletine HCl did not prolong ventricular depolarisation (QRS duration) or repolarisation (QT intervals) as measured by electrocardiography. Theoretically, therefore, Mexiletine HCl may be useful in the treatment of VA associated with a prolonged QT interval.11
In patients with pre-existing conduction defects, depression of the sinus rate, prolongation of sinus node recovery time, decreased conduction velocity and increased effective refractory period of the intraventricular conduction system have occasionally been observed. 11
The antiarrhythmic effect of Mexiletine HCl has been established in controlled comparative trials against placebo, quinidine, procainamide and disopyramide. Mexiletine HCl, at doses of 200-400 mg q8h, produced a significant reduction of ventricular premature beats, paired beats, and episodes of nonsustained VT compared to placebo and was similar in effectiveness to the active agents. Among all the patients that entered into the studies, about 30% in each treatment group had a 70% or greater reduction in PVC count and about 40% failed to complete the 3 month studies because of adverse effects. Follow-up of patients from the controlled trials has demonstrated the continued effectiveness of Mexiletine HCl in long-term use. 11
Hemodynamics
Hemodynamic studies in a limited number of patients, with normal or abnormal myocardial function, following oral administration of Mexiletine HCl, have shown small, usually not statistically significant, decrease in cardiac output and increase in systemic vascular resistance, but no significant negative inotropic effect. Blood pressure and pulse rate remain essentially unchanged. Mild depression of myocardial function, similar to that produced by lidocaine, has occasionally been observed following IV Mexiletine HCl therapy in patients with cardiac disease. 11
Pharmacokinetics
After IV administration of Mexiletine there is extensive tissue uptake with an initial rapid and subsequent slow distribution phase. The volume of distribution is over 600L in healthy subjects. The bioavailability after oral administration in healthy volunteers is about 90%. Peak plasma concentrations are achieved in 2 to 4 hours and there is a linear dose-plasma concentration relationship in the dose range 100 to 600mg. With multiple oral administration steady-state is achieved after 4 or 5 days with no evidence of accumulation.11
There is extensive hepatic metabolism of Mexiletine to inactive metabolites, and only about 8 to 15% of an administered dose is eliminated unchanged in the urine. At physiological pH, the elimination half-life has been reported in the range 7.5 to 11.3 hours, but this is prolonged in the presence of alkaline urine. A sustained release formulation of Mexiletine (360mg capsules) has equivalent bioavailability to the standard preparation but produces lower peak plasma concentrations at 4 to 6 hours after administration and maintains higher trough concentrations. 11
Patients with recent acute myocardial infarction (AMI) have delayed absorption of Mexiletine. They also have an increased volume of distribution and prolonged elimination. In contrast there is very little variation in the pharmacokinetics of Mexiletine in patients with renal disease, even those on dialysis, or in elderly patients. Patients with hepatic disease may have a markedly prolonged elimination half-life and reduced rate of clearance of Mexiletine, resulting in plasma concentrations above the therapeutic range of 0.5 to 2 mg/L in some. 11
Indications and Usage
Mexiletine HCl is indicated for the treatment of documented VA, such as sustained VT, that, in the judgement of the physician, are life-threatening. Because of the proarrhythmic effects of Mexiletine HCl, its use with lesser arrhythmias is generally not recommended. Treatment of patients with asymptomatic ventricular premature contractions should be avoided.11
Initiation of Mexiletine HCl treatment, as with other antiarrhythmic agents used to treat life-threatening arrhythmias, should be carried out in the hospital. Antiarrhythmic drugs have not been shown to enhance survival in patients with VA. 11
Dosage and Administration
The dosage of Mexiletine HCl must be individualised on the basis of response and tolerance, both of which are dose-related. Administration with food or antacid is recommended. Initiate Mexiletine HCl therapy with 200 mg every eight hours when rapid control of arrhythmia is not essential. A minimum of two to three days between dose adjustments is recommended. Dose may be adjusted in 50 or 100 mg increments up or down. 11
As with any antiarrhythmic drug, clinical and electrocardiographic evaluation (including Holter monitoring if necessary for evaluation) are needed to determine whether the desired antiarrhythmic effect has been obtained to guide titration and dose adjustment.
Satisfactory control can be achieved in most patients by 200 to 300 mg given every eight hours with food or antacid. If satisfactory response has not been achieved at 300 mg q8h, and the patient tolerates Mexiletine HCl well, a dose of 400 mg q8h may be tried. As the severity of CNS side-effects increases with total daily dose, the dose should not exceed 1200 mg/day. 11
In general, patients with renal failure will require the usual doses of Mexiletine HCl. Patients with severe liver disease, however, may require lower doses and must be monitored closely. Similarly, marked rightsided congestive heart failure can reduce hepatic metabolism and reduce the needed dose. Plasma level may also be affected by certain concomitant drugs. 11
Loading Dose
When rapid control of ventricular arrhythmia is essential, an initial loading dose of 400 mg of Mexiletine HCl may be administered, followed by a 200 mg dose in eight hours. Onset of therapeutic effect is usually observed within 30 minutes to two hours. 11
Q12H Dosage Schedule
Some patients responding to Mexiletine HCl may be transferred to a 12-hour dosage schedule to improve convenience and compliance. If adequate suppression is achieved on a Mexiletine HCl dose of 300 mg or less every eight hours, the same total daily dose may be given in divided doses every 12 hours while carefully monitoring the degree of suppression of ventricular ectopy. This dose may be adjusted up to a maximum of 450 mg every 12 hours to achieve the desired response. 11
Transferring to Mexohar (Mexiletine HCl)
The following dosage schedule, based on theoretical considerations rather than experimental data, is suggested for transferring patients from other Class I oral antiarrhythmic agents to Mexiletine HCl: Mexiletine HCl treatment may be initiated with a 200 mg dose, and titrated to response as described above, 6-12 hours after the last dose of quinidine sulphate, 3-6 hours after the last dose of procainamide, 6-12 hours after the last dose of disopyramide or 8-12 hours after the last dose of tocainide.11
In patients in whom withdrawal of the previous antiarrhythmic agent is likely to produce life-threatening arrhythmias, hospitalisation of the patient is recommended.
When transferring from lidocaine to Mexiletine HCl, the lidocaine infusion should be stopped when the first oral dose of Mexiletine HCl is administered. The infusion line should be left open until suppression of the arrhythmia appears to be satisfactorily maintained.11
Consideration should be given to the similarity of the adverse effects of lidocaine and Mexiletine HCl and the possibility that they may be additive.
Contraindications
The absolute contraindications to using Mexiletine include untreated second or third-degree heart block, cardiogenic shock, and hypersensitivity to the drug or drug-class.11 It should be used cautiously in a patient with conduction abnormalities that are not second or third-degree heart blocks, or heart failure. Mexiletine is metabolised by the liver and should be cautiously used in any hepatic impairment as it may increase the half-life and risk of toxicity. Electrolyte abnormalities can increase the risk of the development of new arrhythmias and should be used repleted prior to use, especially hypokalaemia.10
Adverse Effects
The adverse effect profile of mexiletine is extensive and limits its use. It affects nearly every organ system, most notably, it has significant cardiovascular and central nervous system (CNS) side-effects, along with affecting the gastrointestinal (GI), musculoskeletal, dermatologic systems. 10
Cardiovascular adverse effects include bradycardia, palpitations, angina, ventricular premature contractions, heart block, hypotension and exacerbation of cardiac arrhythmias. CNS adverse effects include dizziness, ataxia, abnormal gait, numbness, paresthesias, confusion, headache, and tremors. GI adverse effects include the risk of pill-induced esophagitis, nausea, vomiting, constipation, diarrhea, and abdominal pain. The musculoskeletal system can exhibit weakness, arthralgias, and tremors, which is often a pertinent sign when considering the toxicity of Mexiletine. Dermatologic signs include a spectrum of skin rashes, from benign rashes to being a component of Drug Reactions with Eosinophilia and Systemic Symptoms (DRESS) and Stevens-Johnson syndrome. Further severe adverse effects of hepatotoxicity, leukopenia, agranulocytosis, and thrombocytopenia have also been reported in the literature.10
Mexiletine carries a black box warning relative to the findings of the Cardiac Arrhythmia Suppression Trial (CAST) published in 1989, where suppression of asymptomatic, non-life threatening ventricular arrhythmias in post-myocardial infarction patients lead to excessive mortality than the placebo group. Though class 1C antiarrhythmics were used for suppression in the study, the results are extrapolated to across all class 1 antiarrhythmics.10
Warnings
Mortality: In the National Heart, Lung and Blood Institute's Cardiac Arrhythmia Suppression Trial (CAST), a long-term, multicentre, randomised, double-blind study in patients with asymptomatic nonlife-threatening VA who had a myocardial infarction more than six days but less than two years previously, an excessive mortality or non-fatal cardiac arrest rate (7.7%) was seen in patients treated with encainide or flecainide compared with that seen in patients assigned to carefully matched placebotreated groups (3.0%). The average duration of treatment with encainide or flecainide in this study was ten months. The applicability of the CAST results to other populations (e.g., those without recent myocardial infarction) is uncertain. Considering the known proarrhythmic properties of Mexiletine HCl and the lack of evidence of improved survival for any antiarrhythmic drug in patients without life-threatening arrhythmias, the use of Mexiletine HCl as well as other antiarrhythmic agents should be reserved for patients with life-threatening VA.11
Acute Liver Injury
In post-marketing experience abnormal liver function tests have been reported, some in the first few weeks of therapy with Mexiletine HCl. Most of these have been observed in the setting of congestive heart failure or ischemia and their relationship to Mexiletine HCl has not been established.11
Precautions
General
If a ventricular pacemaker is operative, patients with second or third degree heart block may be treated with Mexiletine HCl if continuously monitored. A limited number of patients (45 of 475 in controlled clinical trials) with pre-existing first degree AV block were treated with Mexiletine HCl; none of these patients developed second or third degree AV block. Caution should be exercised when it is used in such patients or in patients with pre-existing sinus node dysfunction or intraventricular conduction abnormalities.
Like other antiarrhythmics Mexiletine HCl can cause worsening of arrhythmias. This has been uncommon in patients with less serious arrhythmias (frequent premature beats or non-sustained VT: See adverse reactions), but is of greater concern in patients with life-threatening arrhythmias such as sustained VT. In patients with such arrhythmias subjected to programmed electrical stimulation or to exercise provocation, 10-15% of patients had exacerbation of the arrhythmia, a rate not greater than that of other agents.
Mexiletine HCl should be used with caution in patients with hypotension and severe congestive heart failure because of the potential for aggravating these conditions. Since Mexiletine HCl is metabolised in the liver, and hepatic impairment has been reported to prolong the elimination half-life of Mexiletine HCl, patients with liver disease should be followed carefully while receiving Mexiletine HCl. The same caution should be observed in patients with hepatic dysfunction secondary to congestive heart failure. Concurrent drug therapy or dietary regimens which may markedly alter urinary pH should be avoided during Mexiletine HCl therapy. The minor fluctuations in urinary pH associated with normal diet do not affect the excretion of Mexiletine HCl.11
SGOT Elevation and Liver Injury
In three-month controlled trials, elevations of SGOT greater than three times the upper limit of normal occurred in about 1% of both Mexiletine-treated and control patients. Approximately 2% of patients in the mexiletine compassionate use programme had elevations of SGOT greater than or equal to three times the upper limit of normal. These elevations frequently occurred in association with identifiable clinical events and therapeutic measures such as congestive heart failure, acute myocardial infarction, blood transfusions and other medications. These elevations were often asymptomatic and transient, usually not associated with elevated bilirubin levels and usually did not require discontinuation of therapy. Marked elevations of SGOT (> 1000 U/L) were seen before death in four patients with end-stage cardiac disease (severe congestive heart failure, cardiogenic shock).
Rare instances of severe liver injury, including hepatic necrosis, have been reported in association with Mexiletine HCl treatment. It is recommended that patients in whom an abnormal liver test has occurred, or who have signs or symptoms suggesting liver dysfunction, be carefully evaluated. If persistent or worsening elevation of hepatic enzymes is detected, consideration should be given to discontinuing therapy. Mexiletine should be used with caution in patients with known seizure disorder.11
Blood Dyscrasias
Among 10,867 patients treated with Mexiletine in the compassionate use programme, marked leukopenia (neutrophils less than 1000/mm3) or agranulocytosis were seen in 0.06% and milder depressions of leukocytes were seen in 0.08%, and thrombocytopenia was observed in 0.16%. Myelofibrosis was reported in two patients in the compassionate use programme: One was receiving long-term thiotepa therapy and the other had pretreatment myeloid abnormalities.
In post-marketing experience, there have been isolated, spontaneous reports of pulmonary changes including pulmonary infiltration and pulmonary fibrosis during Mexiletine therapy with or without other drugs or diseases that are known to produce pulmonary toxicity. A causal relationship to Mexiletine therapy has not been established. In addition, there have been isolated reports of drowsiness, nystagmus, ataxia, dyspepsia, hypersensitivity reaction, and exacerbation of congestive heart failure in patients with pre-existing compromised ventricular function. There have been rare reports of pancreatitis associated with Mexiletine treatment.11
Many of these patients were seriously ill and receiving concomitant medications with known hematologic adverse effects. Rechallenge with Mexiletine in several cases was negative. Marked leukopenia or agranulocytosis did not occur in any patient receiving Mexiletine HCl alone; five of the six cases of agranulocytosis were associated with procainamide (sustained release preparations in four) and one with vinblastine. If significant hematologic changes are observed, the patient should be carefully evaluated, and, if warranted, Mexiletine HCl should be discontinued. Blood counts usually return to normal within one month of discontinuation.
Convulsions (seizures) did not occur in Mexiletine HCl controlled clinical trials. In the compassionate use programme, convulsions were reported in about 2 of 1000 patients. Twenty-eight percent of these patients discontinued therapy. Convulsions were reported in patients with and without a prior history of seizures. Mexiletine should be used with caution in patients with known seizure disorder. 11
Carcinogenesis, Mutagenesis and Impairment of Fertility
Studies of carcinogenesis in rats (24 months) and mice (18 months) did not demonstrate any tumorigenic potential. Mexiletine HCl was found to be non-mutagenic in the Ames test. Mexiletine HCl did not impair fertility in the rat.11
Pregnancy
Teratogenic Effects - Pregnancy Category C. Reproduction studies performed with Mexiletine HCl in rats, mice and rabbits at doses up to four times the maximum human oral dose (24 mg/kg in a 50 kg patient) revealed no evidence of teratogenicity or impaired fertility but did show an increase in foetal resorption. There are no adequate and well-controlled studies in pregnant women; this drug should be used in pregnancy only if the potential benefit justifies the potential risk to the foetus.11
Nursing Mothers
Mexiletine HCl appears in human milk in concentrations similar to those observed in plasma. Therefore, if the use of Mexiletine HCl is deemed essential, an alternative method of infant feeding should be considered.11
Paediatric Use
Safety and effectiveness in the paediatric population have not been established. 11
Guideline Recommendations
Select recommendations from the 2017 American Heart Association (AHA)/American College of Cardiology (ACC)/Heart Rhythm Society (HRS) guideline for the management of patients with VA and the prevention of sudden cardiac death include the following7:
- Mexiletine is indicated in the use in VT, VF, PVC, has a role in patients with Long QT syndrome 3 (LQT3) with no marked effect on most intervals
Recommendations from the 2015 European Society of Cardiology (ESC) guideline for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death:
- Mexiletine is indicated in the treatment of VT, LQT 3
- Sodium channel blockers (mexiletine, flecainide or ranolazine) may be considered as add-on therapy to shorten the QT interval in LQTS3 patients with a QTc >500 ms.
Recommendations from the 2014 European Heart Rhythm Association (EHRA)/ Heart Rhythm Society (HRS)/ Asia Pacific Heart Rhythm Society (APHRS) expert consensus on VA:
- In patients who suffer from symptomatic non-sustained VAs on an adequately dosed beta-blocker or an on-dihydropyridine calcium channel antagonist, treatment with an antiarrhythmic drug (AAD; amiodarone, flecainide, mexiletine, propafenone, sotalol) maybe considered to improve symptoms associated with arrhythmia episodes (Class IIb LOE C)
- For patients with SHD and recurrent SMVT, specific treatment of Vas with AADs (amiodarone, mexiletine, or sotalol), catheter ablation, and/or antitachycardia pacing (ATP) from an ICD should be considered in addition to an ICD (Class IIa LOE B)
Clinical Evidence for Mexiletine
Mexiletine for Control of Drug-resistant VT12
Aim:
To evaluate the antiarrhythmic efficacy of Mexiletine patients with drug-resistant ventricular tachyarrhythmias.
Study Design:
33 patients with drug-resistant ventricular tachyarrhythmias. The efficacy of Mexiletine was assessed on the basis of the results of programmed ventricular stimulation.
Result:
Mexiletine did not alter the ventricular effective refractory period, the Q-Tc interval, or the methods of tachyarrhythmia induction and termination during programmed stimulation
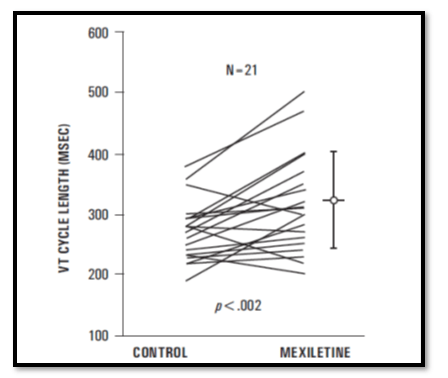
The mean cycle length of VT increased from 270 +/- 49 to 313 +/- 80 ms in 21 patients in whom VT remained inducible on Mexiletine alone (p<0.002) Overall, VT remained inducible with methods similar to control (no drugs) inductions in 25 patients receiving Mexiletine alone or in combination with a type I agent
VT induction was prevented in only 8 patients, 3 on Mexiletine alone and 5 receiving Mexiletine combined with another drug
 /a>
/a>Efficacy of Mexiletine in Chronic VA compared with Quinidine: A Single-Blind, Randomised Trial.13
There was no significant difference in the 2 groups in side-effects.
Clinical Evaluation of Oral Mexiletine Therapy in the Treatment of VA4
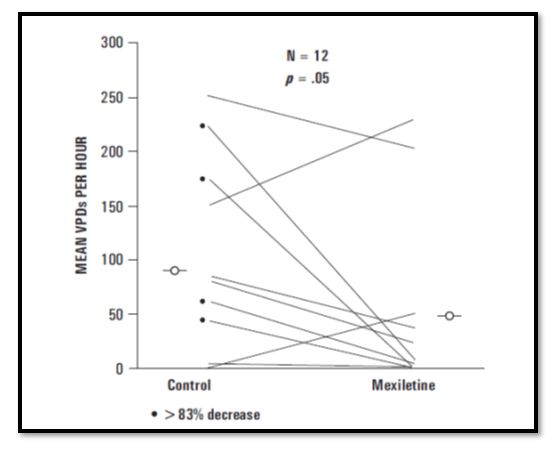
19 patients (36.5%) had a greater than 83% decrease in ventricular ectopic rhythm.
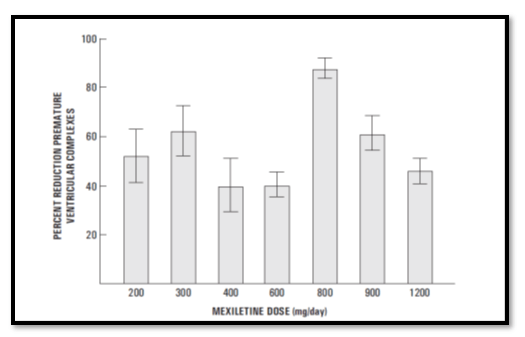
Mexiletine was effective in a modest proportion of patients in whom conventional antiarrhythmic therapy had failed. Ventricular function appeared to be unaffected by the drug and the side-effects, although relatively frequent, were not serious.
Mexiletine in the Treatment of Resistant VA: Enhancement of Efficacy and Reduction of Dose-related Side-Effects by Combination with Quinidine15
Aim:
To treat patients whose VA were not responsive to conventional antiarrhythmic therapy (or in whom therapy produced limiting side-effects) with Mexiletine.
Study Protocol:
21 patients
Results:
In the group of 17 patients, the addition of a previously well-tolerated dosage of quinidine (824 + 298 mg) to a well-tolerated but only partially effective dosage of Mexiletine (800 + 239 mg) produced a significantly greater antiarrhythmic response.
The mean suppression of ventricular ectopic depolarisation (VEDs) during combination therapy increased to 85.9 + 26%.
Only one patient continued to have VT, and limiting side-effects occurred in only 12% of the patients.
Continuation of quinidine and withdrawal of Mexiletine was associated with recurrence of complex VA and documented the need for combination treatment in nine patients.
The coupling interval of the predominant ectopic beat prolonged (p<0.05) during Mexiletine treatment and further prolonged (p< 0.05) with the addition of quinidine.
After withdrawal of Mexiletine from the combination treatment in 9 patients, the QTc interval significantly prolonged (p<0.05). Thus, Mexiletine limited the quinidine-induced increase in QTc interval.
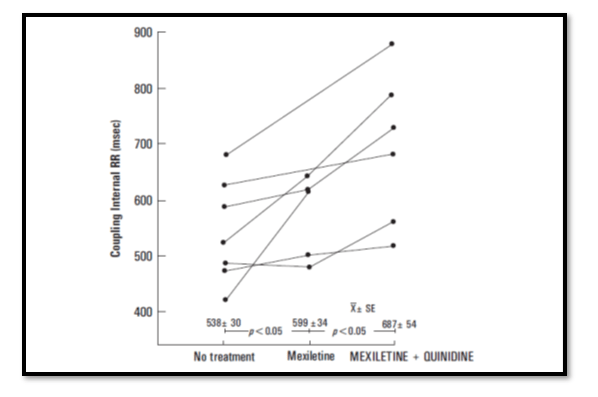
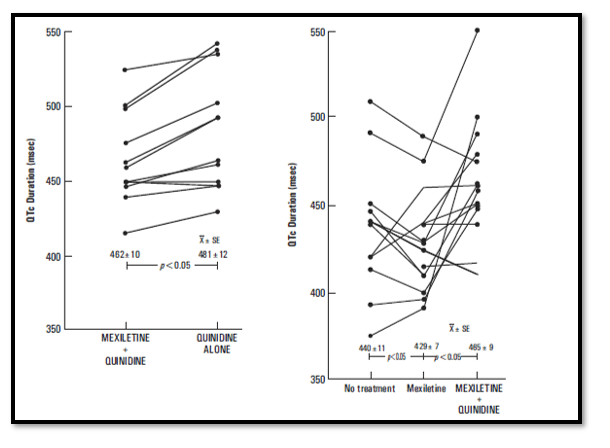
During 18 ± 5.2 months of follow-up, three patients have died, one with intractable congestive heart failure and two related to documented noncompliance to their medical regimen.
- The addition of quinidine, which prolongs repolarisation of the action potential in vitro, enhanced the antiarrhythmic efficacy of Mexiletine, which shortens the action potential duration in vitro.
Conclusion:
The authors conclude that combinations of drugs with different electrophysiologic properties may provide enhanced antiarrhythmic efficacy.
Mexiletine in Treatment of VA in Patients with Recent Myocardial Infarction16
Aim:
The aim of the study was to investigate the antiarrhythmic effect of oral Mexiletine in patients at high risk after acute myocardial infarction
Study Protocol:
This was a double-blinded trial enrolling a total of 344 patients at high risk after acute myocardial infarction. Mexiletine was administered in an initial loading dose of 400 mg, followed by 250 mg 8 hourly; 3 months
Result:
- A statistically significant reduction in incidence of couplets was observed at 1 month was observed (34% Mexiletine, 48% placebo)
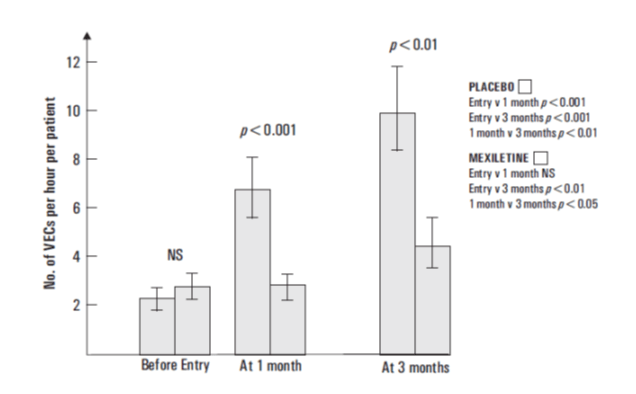
In the placebo group the prevalence was significantly higher at 1 month than at entry and again at 3 months compared with 1 month. The prevalence was significantly reduced by Mexiletine.
- Potential unwanted effects of therapy, particularly those related to the CNS and the GI tract, were more common in patients treated with Mexiletine than in those on placebo. Side-effects led to 30 withdrawals in the Mexiletine group and 6 in the placebo group. There was no evidence that Mexiletine caused any important CV side-effects. 24 patients (13%) in the Mexiletine group and 19 (12%) in the placebo group died.
Conclusion:
The authors concluded that Mexiletine reduced the prevalence of VA in a high-risk group of patients with recent myocardial infarction. The authors also mention that no favourable effect on mortality was observed.
Mexiletine in Treatment of Patients with Recurrent Ventricular Tachyarrhythmias and ICD Shocks17
Aim:
The aim of this study was to evaluate the efficacy and tolerability of Mexiletine in patients with recurrent ventricular tachyarrhythmias and/or electrical storm events, in whom standard treatment strategies failed to prevent ventricular tachyarrhythmia.
Study Protocol:
A total of 17 patients with recurrent VT and/or VF were included in the study and the Mexiletine dose given was 600 mg/day in 13 patients and 400 mg/day in four patients. The Mexiletine treatment was given for 8 months.
Results:
- Treatment with Mexiletine in comparison with a matched period before initiation of treatment significantly reduced the number of:
- Electrical storm events (14 episodes vs. 2 episodes; median and IR for 17 patients: 1 [0–1] vs. 0 [0–0], p = 0.0010)
- VT/VF episodes (285 vs. 74 episodes; median and IR for 17 patients: 7 [5–27] vs. 0 [0–5], p = 0.0115)
- ICD interventions (317 interventions vs. 9 interventions; median and IR for 17 patients: 10 [5–25] vs. 0 [0–2], p = 0.0006)
- In 14 out of 17 patients (82%) sufficient tolerability of Mexiletine was observed. Only in three (18%) patients severe side-effects of Mexiletine treatment occurred requiring discontinuation of therapy.
Conclusion:
The authors concluded that Mexiletine may be effective in the treatment of recurring ventricular tachyarrhythmias or electrical storm events.
Conclusion
Mexiletine, a Class IB antiarrhythmic has been on the market for several years and has shown safety and efficacy as a stable molecule through many in vivo and ex vivo studies. Despite, the fact that most of the studies performed on Mexiletine were in the 20th century, the stability of the molecule is unquestioned through several systematic reviews and a positive clinical experience in patients, therefore, often making it the choice of drug prescribed by physicians.
References
- Qu Z and Weiss JN. Mechanisms of ventricular arrhythmias from molecular fluctuations to electrical turbulence. Annu Rev Physiol. 2015; 77:29–55.
- Flinders D. C. and Roberts, S. D. Ventricular Arrhythmias. Primary Care: Clinics in Office Practice. 2000;27 (3):709–724
- Arrhythmia.Available at: https://www.nhlbi.nih.gov/health-topics/arrhythmia. Last accessed on: 24-05-2019
- Vora A, Naik A, Lokhandwala Y, et al. Profiling cardiac arrhythmia and heart failure patients in India: The Panarrhythmia and Heart Failure Observational Study. Indian Heart J. 2017;69(2):226-239.
- Steven J Compton. Ventricular Tachycardia. Available at: https://emedicine.medscape.com/article/159075overview#a6. Last accessed on: 24-05-2019.
- Steven J Compton. Which rhythms must be distinguished from ventricular tachycardia (VT). Available at: https://www.medscape.com/answers/159075-67691/which-rhythms-must-be-distinguished-fromventricular-tachycardia-vt Last accessed on: 24-05-2019.
- Steven J Compton. Approach Considerations. Available at: https://emedicine.medscape.com/article/159075treatment. Last accessed on: 24-05-2019.
- Steven J Compton. Ventricular Tachycardia Treatment & Management. Available at: https://emedicine. medscape.com/article/159075-treatment#d8 Last accessed on: 24-05-2019.
- Steven J Compton. Ventricular Tachycardia Treatment & Management. Available at: https://emedicine.medscape.com/article/159075-treatment#d14 Last accessed on: 24-05-2019.
- Singh S, Zeltser R. Mexiletine. In: StatPearls Treasure Island (FL): StatPearls Publishing; 2019 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK519045/
- Mexiletine Hydrochloride Prescribing Information. Available at: https://docs.boehringer-ingelheim.com/ Prescribing%20Information/PIs/Mexitil%20Caps/Mexitil.pdf
- Waspe LE, Waxman HL, Buxton AE, et al. Mexiletine for control of drug-resistant ventricular tachycardia: Clinical and electrophysiologic results in 44 patients. The American Journal of Cardiology. 1983;51 ( 7):1175–1181
- Singh JB, Rasul AM, Shah A, et al. Efficacy of mexiletine in chronic ventricular arrhythmias compared with quinidine: A single-blind, randomised trial. The American Journal of Cardiology. 1984; 53(1) 84–87
- Rutledge JC, Harris F and Amsterdam EA. Clinical evaluation of oral mexiletine therapy in the treatment of ventricular arrhythmias. J Am Coll Cardiol. 1985;6(4):780-4.
- Duff HJ, Roden D, Primm RK, et al. Mexiletine in the treatment of resistant ventricular arrhythmias: enhancement of efficacy and reduction of dose-related side-effects by combination with quinidine. Circulation.1983;67 (5):1124-8.
- Chamberlain DA, Jewitt DE, Julian DG, et al. Oral mexiletine in high-risk patients after myocardial infarction. Lancet. 1980; 20-27;2 (8208-8209):1324-7.
- Sobiech M, Lewandowski M, Zajac D, et al. Efficacy and tolerability of mexiletine treatment in patients with recurrent ventricular tachyarrhythmias and implantable cardioverter-defibrillator shocks. Kardiol Pol. 2017;75(10):1027-1032.