Monograph
Endobloc (Ambrisentan 5 Mg /10 Mg) PAH Management Simplified
Pulmonary hypertension is an observation, not a single diagnosis or disease. It encompasses a diverse group of
conditions that lead to elevated pulmonary pressures. It is defined clinically as an increase in the pulmonary
vascular pressure that is caused by conditions that are associated with an increase in the pulmonary arterial
pressure or both the arterial and venous pressure. Haemodynamically, it is defined as an increase in
the
mean pulmonary arterial pressure to >25 mmHg at rest or >30 mmHg during exercise.
Pulmonary hypertension was previously classified as either primary or secondary pulmonary hypertension, depending
on the absence or presence of identifiable causes of increased pulmonary pressure. The 2008 World Symposium on
Pulmonary Hypertension set forth a new classification system that categorizes pulmonary hypertension on the
basis of the following clinical criteria:
Updated clinical classification of pulmonary hypertension (Dana Point, 2008)
1. Pulmonary arterial hypertension (PAH) 2. Pulmonary hypertension due to left heart disease 3. Pulmonary
hypertension due to lung diseases and/or hypoxia 4. Chronic thromboembolic pulmonary hypertension 5. Pulmonary
hypertension with unclear and/or multifactorial mechanisms
PAH (World Health Organization [WHO] Group I) is defined as a sustained elevation of
pulmonary arterial pressure to >25 mmHg at rest or to >30 mmHg with exercise, with a mean
pulmonary-capillary wedge pressure and left ventricular end-diastolic pressure of <15 mmHg.
PAH
is a life-threatening and rare condition characterized by vasoconstriction and vascular remodelling, resulting in a
progressive increase in pulmonary vascular resistance (PVR) and pulmonary artery pressure (PAP), leading to right
heart failure and death. Its prevalence is estimated at 15 cases per million people each year, with 2-3 times as
many women versus men afflicted. PAH may be idiopathic (IPAH), familial PAH (FPAH) or PAH associated (APAH) with
various conditions, such as connective tissue disease, congenital systemic-to-pulmonary shunts, portal hypertension,
drug and toxin use, and HIV infection. PAH comprises a group of heterogeneous conditions that share comparable
clinical and haemodynamic features and virtually identical pathological changes of the microcirculation of the
lungs. The prevalence varies substantially depending on the type, aetiology and underlying condition.

The diagnosis of PAH should be considered in any patient with unexplained dyspnoea on
exertion,fatigue or exercise limitation, those with clinical signs consistent with right-heart dysfunction (e.g.,
peripheral oedema, ascites), and patients with symptoms and having a process known to be associated with PAH and/or
a family history of pulmonary hypertension. The initial assessment of PAH is to determine whether pulmonary
hypertension is present, with a non-invasive evaluation (screening echocardiogram). If there is evidence of
pulmonary hypertension, then an invasive method (right-heart catheterization) is required to confirm this finding
and evaluate if its haemodynamically consistent with PAH (i.e., increased pulmonary vascular resistance with normal
wedge pressure). This method is also used to evaluate the severity of the disease. The main vascular changes in PAH
are vasoconstriction, smooth muscle cell and endothelial cell proliferation, and thrombosis. Advances in the
understanding of the molecular mechanisms involved in this disease suggest that endothelial dysfunction plays a key
role. Chronically impaired production of vasoactive mediators, such as nitric oxide and prostacyclin, along with
prolonged over-expression of vasoconstrictors such as endothelin-1 (ET-1), not only affect vascular tone but also
promote vascular remodelling. Thus, these substances represent logical pharmacological targets. First-line treatment
comprises general/supportive care, including supplemental oxygen (O2), diuretics, oral anticoagulants and avoidance
of exacerbations. Patients should also be evaluated via acute vasoreactivity testing to determine if they are likely
to respond to oral calcium channel blockers. Patients who are not candidates for calcium channel blockers therapy or
have not responded to it have a variety of treatment options depending on the severity of their disease. The options
available are prostacyclin analogues, ET-receptor antagonists (ETRAs) and phosphodiesterase type 5 (PDE-5)
inhibitors. Although none of them cure this devastating condition, the treatment options for patients with PAH have
evolved, helping to prevent disease progression, prolong patient survival and improve their quality of life.
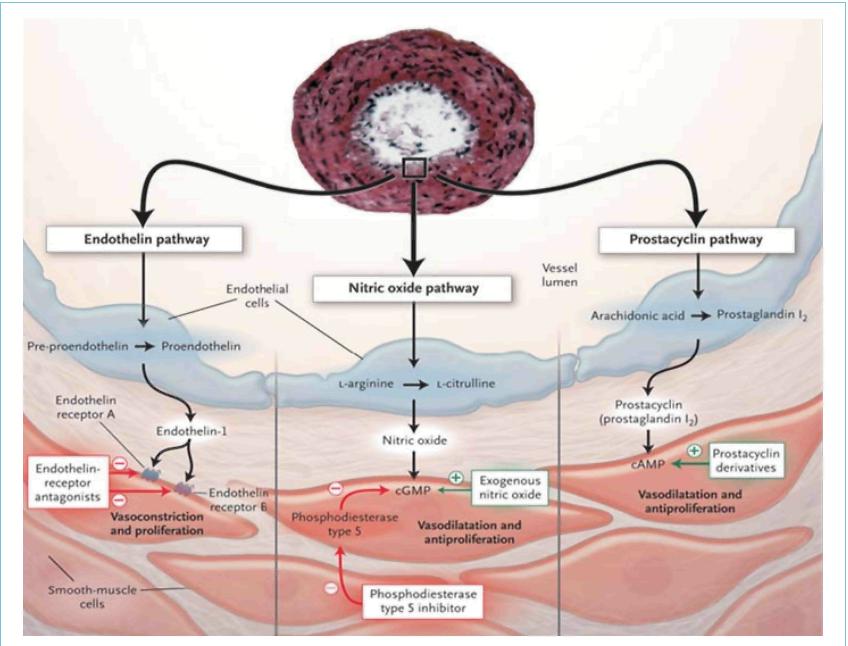
ET-1, a peptide produced primarily by vascular endothelial cells, is characterized as a powerful
vasoconstrictor and mitogen for smooth muscle. ET-1 binds to two types of receptors, ETA and
ETB; ETA- receptors are found in smooth muscle cells whereas ETB-receptors are localized on both the
endothelial cells and in smooth muscle cells. Activation of ETA- and ETB-receptors on smooth muscle cells mediates
the vasoconstrictive and mitogenic effects of ET-1. Stimulation of ETB-receptors promotes ET-1 clearance and
activation of nitric oxide and prostacyclin release. Currently, three different ETRAs are available. Bosentan, the
first drug in this category is a dual ETRA whereas ambrisentan and sitaxsentan selectively block only the
ETA-receptors.
These drugs are selective inhibitors of cyclic guanylate monophosphate (cGMP) PDE-5 that exert its
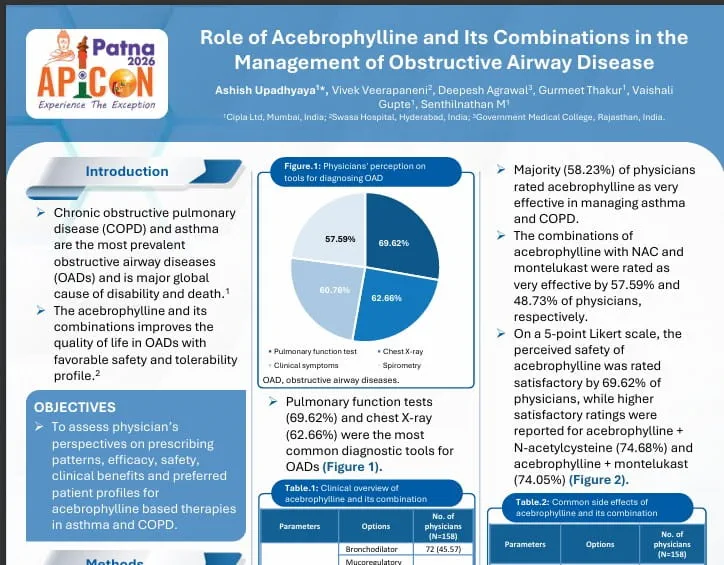
pharmacological effect by increasing the intracellular concentration of cGMP. The increase of this nucleotide
induces relaxation and antiproliferative effects on the vascular smooth muscle cells. PDE-5 is selectively abundant
in the pulmonary circulation and PDE-5 gene expression and activity are increased in chronic pulmonary hypertension.
Sildenafil and tadalafil are the PDE-5 inhibitors approved for use in PAH.
These drugs act mainly by relaxing the vascular smooth muscle cells (acute) and inhibiting platelet
aggregation; however, the precise mechanism of action of prostacyclin administration in PAH is unknown and is likely
to be multifactorial. Though these drugs have contributed tremendously to the management of PAH, their usage has
been limited due to drawbacks such as the route of administration, frequent dosing schedules and short half-lives.
However, PAH is a complex disorder and targeting a single pathway cannot be expected to be uniformly successful.
Thus, combining substances with different modes of action is expected to improve the symptoms, haemodynamics and
survival rates in PAH patients.
The ETs are a family of 21 amino acid peptides produced by the vascular endothelial cells.
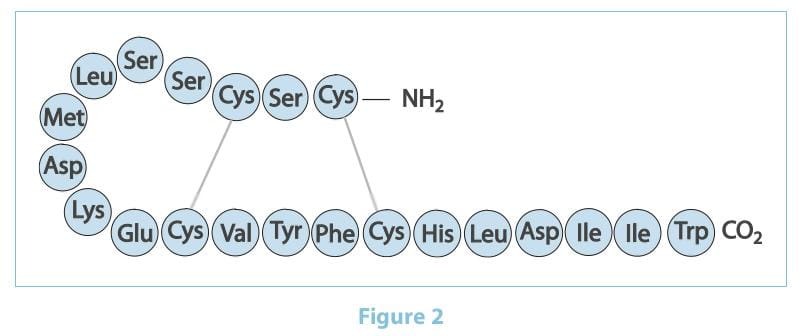
There are three ET isoforms termed ET-1, ET-2 and ET-3, which are encoded by three distinct genes.
ET-1 is considered the predominant and more important pathophysiological isoform. It was first isolated in 1988 by
Yanagisawa as the most potent vasoconstrictor ever identified. It is generated through the cleavage of prepro-ET-1
to big ET-1 and then to ET-1 by the action of ET-converting enzymes. Pre-clinical studies suggest that the lungs are
the principal production site of ETs, with mRNA expression levels 5 times higher than any other organ studied. It is
predominantly produced by the vascular endothelium and, to a lesser extent, by other cell types, including the
pulmonary artery smooth muscles and the lung fibroblasts. The biosynthesis of ET is triggered by hypoxia, growth
factors, cytokines, shear stress, thrombin and angiotensin II. The lungs not only produce but also clear plasma ET
from the circulation. The ETs are 100 times more potent than noradrenaline and 10 times more potent than angiotensin
II.
Within the mammalian cardiovascular system, ET-1 acts through two receptor subtypes ETA
and ETB. In the vasculature, ETA-receptors are located on smooth muscle cells and fibroblasts,
whereas ETB-receptors are predominantly localized on ET cells and, to a lesser extent, on smooth muscle
cells, fibroblasts and macrophages. Recent data using cultured transfected cell lines suggest that ETA-
and ETB-receptors can form constitutive heterodimers (dimerization theory). Functionally, this means that
ETB-receptors expressed on smooth muscle cells couple with ETA-receptors and the former adopt
the function of the latter, such that ETB-receptors in heterodimers mediate vasoconstriction similar to
ETA-receptors. Furthermore, it has been suggested that selective antagonism of one ET-receptor subtype
only may result in compensation by the other receptor. This experimental hypothesis has been called
'cross-talk'.
- ET-Receptor Selectivity and its Vasoconstriction and Vasodilation Effects
Vasodilation is an important goal of therapeutic intervention for PAH. Theoretically, selective
ETARAs should be more effective in achieving this than non-selective ETARAs/ETBRAs,
given the role played by ETB-receptors in both vasodilation and ET-1 clearance. In animal models of PAH,
however, positive dilatory effects have been observed with both selective ETA-receptor blockade and
non-selective antagonism. Since direct evaluation of the pulmonary circulation requires invasive procedures, the
majority of the available data are extrapolated from human studies performed on blood vessels in the systemic
circulation. Collectively, these studies indicate that (i) selective ETA-receptor blockade results in a
robust vasodilator response and increased blood flow; and, (ii) selective ETB-receptor blockade results
in vasoconstriction and reduced blood flow.
- ET-Receptor Selectivity and Fibrosis
Extra-vascular anti-mitotic and anti-fibrotic effects of ETRAs may result in greater efficacy in
scleroderma than therapies directed exclusively at the vasculature. Data from animal models using either
ETA-selective or non-selective ETRAs demonstrate an amelioration of ET-1-related effects involving the
reduction of the growth factor expression, extracellular matrix deposition and matrix metalloproteinase activity.
Subsequent in vitro data using lung fibroblasts indicate that ET-1 induces collagen matrix contraction
through the ETA-receptor, but not the ETB-receptor. Furthermore, while there is evidence that
ETB- receptors are linked to collagen production in vitro, in vivo animal data with
ETARAs have shown that they effectively block the accumulation of collagen I, III and IV, normalize
pro-collagen I and III mRNA, and abolish the effect of ET-1 on pro-collagen metabolism. Likewise, although there is
evidence that under certain conditions ET-1 can act as a mitogen in vitro through both ETA- and
ETB-receptor activation, ETB-receptors have been shown to inhibit vascular smooth muscle cells
proliferation in vivo. Under normal physiological conditions, the receptor types have broadly opposing
functions. Activation of ETA-receptors mediates vasoconstriction, proliferation, hypertrophy, cell
migration and fibrosis, whereas activation of ETB-receptors stimulates the release of the potent
vasodilators (nitric oxide and prostacyclin), which exhibit anti-proliferative properties and prevents apoptosis.
Importantly, ETB-receptors on the ET cells mediate the clearance of circulating ET-1 in the lungs,
kidneys and liver, with up to 50% of mature ET-1 in healthy subjects and 40% in patients with PAH cleared via the
pulmonary ETB-receptors. ET cell ETB-receptor activation also inhibits ET-converting enzyme-1,
the enzyme that is required to produce mature ET-1. Alterations in the distribution and number of ETA-
and ETB-receptors in conditions such as PAH suggest that their roles in the disease state may differ from
those in normal physiology. For example, there are more ET-1-binding sites in the distal pulmonary vessels of
patients with PAH, and ETB-receptors are also up regulated. ETB-receptors may not exclusively
mediate pulmonary vasodilatation. Because of the effects of a sub-population of ETB-receptors located on
the smooth muscle cells and fibroblasts, the spectrum of possible adverse effects of ETB-receptor
stimulation in patients with pulmonary hypertension includes the induction of vasoconstriction, proliferation and
fibrosis.
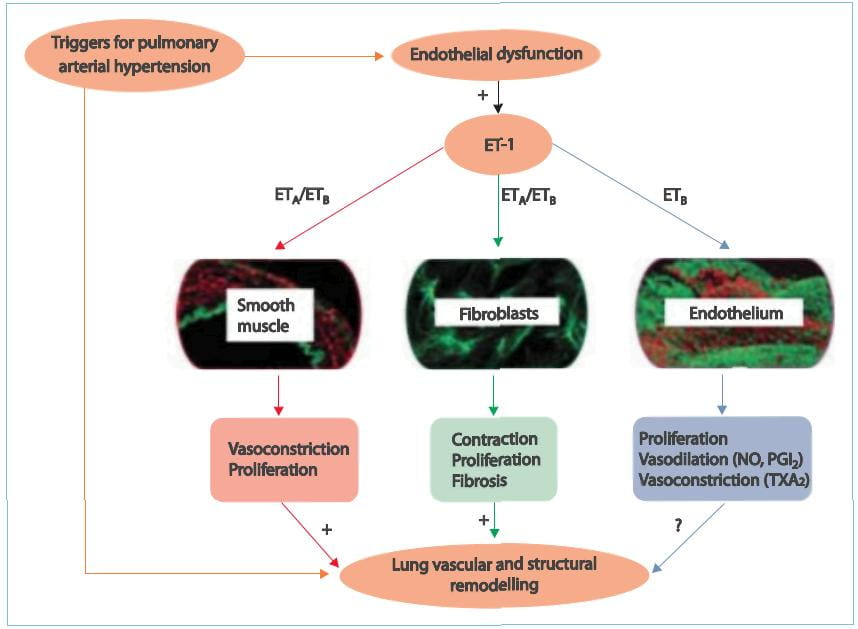
There are studies that suggest that increased ET concentration in the plasma and lung tissue serves
as an important stimulus for sustained pulmonary vasoconstriction and excessive vascular remodelling in patients
with PAH. Selective blockade of the ETA-receptor that mediate contractile and mitogenic effects on
pulmonary arterial smooth muscle cells, while maintaining the function of the ETB-receptor that cause
vasodilative effects and induce ET-1 clearance, is therefore a good strategy for designing therapeutic approaches
for patients with PAH and may offer more benefits than non-selective ETARAs/ETBRAs. Therefore,
there is a potential advantage to use a selective ETRA antagonist such as ambrisentan, which will selectively block
the ETA-receptors, thereby sparing the vasodilative and anti-proliferative action of the
ETB-receptors. The clinical advantage of this is yet to be determined.
In June 2007, the US Food and Drug Administration (FDA) approved ambrisentan for the once-daily
treatment of PAH to improve exercise capacity and delay clinical worsening. Ambrisentan, an orally active, highly
selective antagonist of the ET-1 type A receptor, is indicated for the treatment of PAH. It has a low potential for
drug drug interactions and requires only once-daily administration. Treatment for 3 months with ambrisentan 2.5-10
mg/day significantly improved the exercise capacity, as determined by the distance walked in 6 minutes (6MWD;
primary outcome measure), compared with placebo, in two double-blind, multicentre studies in patients with PAH. A
decrease in dyspnoea and a delay in clinical worsening were among the improvements in secondary outcomes generally
observed with ambrisentan versus placebo. All available pre-registration and postmarketing data indicate that the
drug poses only a very low risk of liver injury; hence, the 'black box' warning regarding potential liver
injury has been removed.
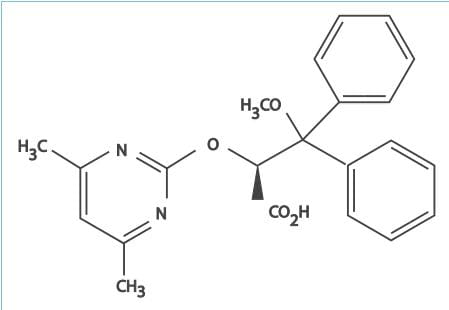
- Ambrisentan is an orally active, diphenyl propionic acid derivative.
- It is a potent antagonist of the ETA-receptor, and has a >4,000-fold higher selectivity for
the ETA-receptor than the ETB-receptor. The ETA-receptors are located
predominantly on vascular smooth muscle cells and mediate vasoconstriction and cell proliferation, whereas
the ETB-receptors are found on both endothelial and vascular smooth muscle cells and primarily
mediate vasodilation, antiproliferation and ET-1 clearance.
- Cardiopulmonary haemodynamic parameters, as assessed by right-heart catheterization, were significantly improved
in patients with PAH who were treated (short- or long-term) with ambrisentan in clinical trials. Ambrisentan
produced sustained, clinically relevant improvements in the mPAP (decreased by 8.2 mmHg from baseline [50.8
mmHg]), PVR (reduced by 297 dyn/s/cm-5 from baseline [856 dyn/s/cm-5]) and the cardiac index (increased by 0.5
L/min/m2 from baseline [2.5 L/min/m2]).
- Right-heart catheterization data were analysed post hoc for a subset of 58 patients with moderate PAH who
received ambrisentan 2.5, 5, or 10 mg once daily in the 2-year, open-label, uncontrolled extension (ARIES-E) of
the 12-week ARIES (Ambrisentan in Pulmonary Arterial Hypertension, Randomized, Double-Blind, Placebo-Controlled,
Multicenter Efficacy) studies. Baseline right-heart catheterization data were assessed at various intervals
before the first dose of ambrisentan (median: 1.4 months); follow-up right-heart catheterization data were
collected at various intervals after the first dose of ambrisentan (median: 13.5 months). Ambrisentan produced
sustained, clinically relevant improvements in the mPAP (decreased by 8.2 mmHg from baseline [50.8 mmHg]), PVR
(reduced by 297 dyn/s/cm-5 from baseline [856 dyn/s/cm-5]) and the cardiac index (increased by 0.5 L/min/m2 from
baseline [2.5 L/min/m2]).
- Right-heart catheterization data were also evaluated retrospectively for a subset of 12 patients with PAH (from
a single institution) who participated in the ARIES-1 and ARIES-E studies. Significant improvements in the
median mPAP, PVR and cardiac output were seen after 1 year of follow-up (all P=0.03 versus baseline); the
improvement in PVR persisted after 2 years of follow-up.
- Oral ambrisentan is rapidly absorbed, with the maximum plasma concentration (Cmax) reached at a
median of ≈ 1.5-2 hours post-dose.
- The absolute bioavailability of ambrisentan is unknown. Ambrisentan is 99% bound to plasma proteins and
accumulates slightly at the steady state.
- The distribution of ambrisentan into red blood cells is low, with a mean blood to plasma ratio of 0.57 and
0.61 in males and females, respectively.
- The pharmacokinetics of ambrisentan is not affected to a clinically significant extent by food.
- The main route of ambrisentan metabolism is glucuronidation to form ambrisentan glucuronide.The drug also
undergoes oxidative metabolism (by cytochrome P450 [CYP450] 3A4 and, to a lesser extent, CYP3A5 and CYP2C19)
to form 4-hydroxymethyl ambrisentan, which is further glucuronidated to 4-hydroxymethyl ambrisentan
glucuronide.
- The main metabolite, 4-hydroxymethyl ambrisentan, has a 64-fold lower binding affinity than the parent
compound for the ETA receptor and is not pharmacologically active.
- The elimination of ambrisentan (and its metabolites) is predominantly by non-renal pathways, with 66% of an
orally administered dose being recovered in the faeces and 22.6% in the urine.
- In patients with PAH, the mean oral clearance of ambrisentan is 19 mL/min. The mean terminal elimination
half-life at the steady state was 15 and 13 hours with ambrisentan dosages of 5 and 10 mg/day, respectively.
- Dosage adjustment is not needed in patients aged >65 years.
- The results of a population pharmacokinetic analysis in patients with PAH and creatinine clearance between
20 and 150 mL/min suggest that mild or moderate renal impairment has no significant influence on exposure to
ambrisentan; hence, dosage adjustment is not required in these populations. However, there is limited or no
experience with the drug in patients with severe renal impairment (creatinine clearance <30 mL/min).
- The influence of hepatic impairment on the pharmacokinetics of ambrisentan has not been determined.
Ambrisentan is not recommended in patients with moderate or severe hepatic impairment. The drug should not
be initiated in patients with severe hepatic impairment or with clinically significant elevated hepatic
aminotransferases (>3 times the upper limit of normal [>3 ULN]).

ARIES-1 and ARIES-2 were concurrent, double-blind, placebo-controlled studies that randomized 202
and 192 patients with PAH, respectively, to placebo or ambrisentan (ARIES-1, 5 or 10 mg; ARIES-2, 2.5 or 5 mg)
orally once daily for 12 weeks. The primary endpoint for each study was as below:
- Change in the 6MWD from baseline to week 12 2. Clinical worsening 3. WHO functional class 4. Short Form-36
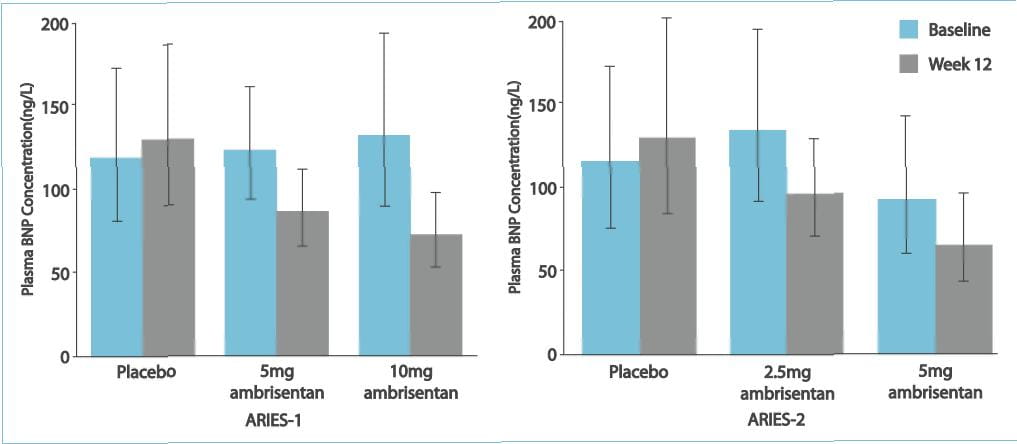
health survey score 5. Borg dyspnoea score 6. B-type natriuretic peptide (BNP) plasma concentrations
In addition, a long-term extension study was performed (48 weeks).
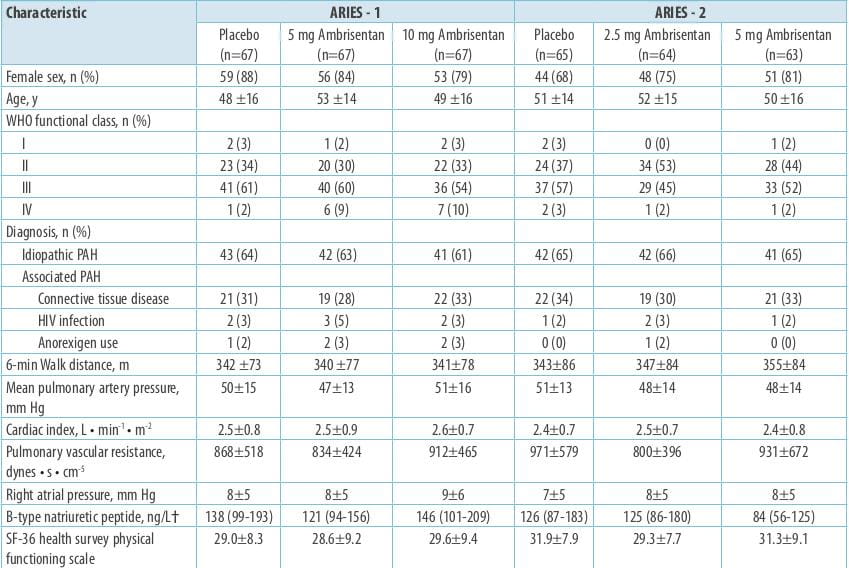
Results
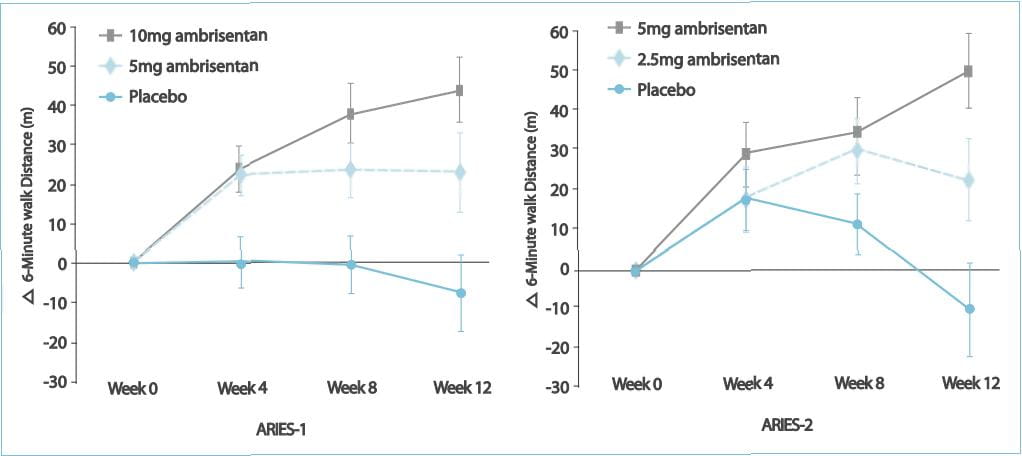
The 6MWD increased in all the ambrisentan groups; mean placebo-corrected treatment effects were 31
metres (P=0.008) and 51 metres (P<0.001) in ARIES-1 for 5 and 10 mg ambrisentan, respectively, and 32 metres
(P=0.022) and 59 metres (P<0.001) in ARIES-2 for 2.5 and 5 mg ambrisentan, respectively.
Improvements in time to clinical worsening (ARIES-2), WHO functional class (ARIES-1), Short Form-36
score (ARIES-2), Borg dyspnoea score (both studies), and BNP (both studies) were observed.
No patient treated with ambrisentan developed aminotransferase concentrations >3xULN. In 280
patients completing 48 weeks of treatment with ambrisentan monotherapy, the improvement from baseline in the 6MWD at
48 weeks was 39 metres. Thus, it was observed that ambrisentan improves the exercise capacity in patients with PAH.
Improvements were observed for several secondary end-points in each of the studies. Ambrisentan is well tolerated
and is associated with a low risk of aminotransferase abnormalities.
This study evaluated the safety and efficacy of ambrisentan for a period of 2 years in patients
with PAH. In the ARIES-1 and ARIES-2 studies, and the subsequent long-term extension protocol, the ARIES-E study,
383 patients received ambrisentan (2.5, 5 or 10 mg).
Results
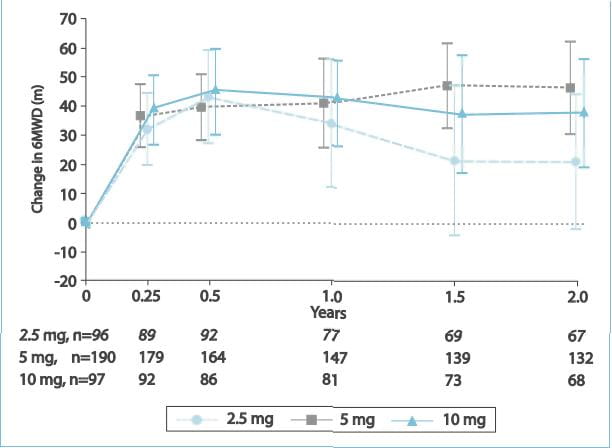
After 2 years of ambrisentan exposure, the mean change from baseline in the 6MWD was improved for
the 5 mg (+23 metres; 95% confidence interval [CI]: 9 to 38 metres) and 10 mg (+28 metres; 95% CI: 11 to 45 metres)
groups.
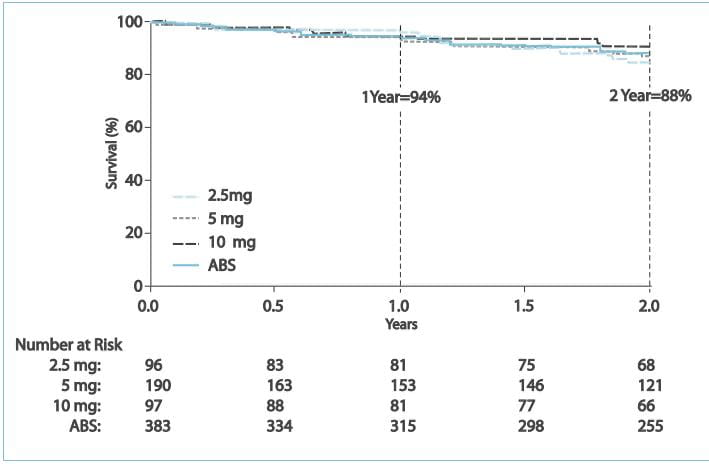
Estimates of survival and freedom from clinical worsening for the
combined dose group were 94% and 83%, respectively, at 1 year and 88% and 72%, respectively, at 2 years.
The annualized risk of aminotransferase (alanine aminotransferase [ALT] and/or aspartate
aminotransferase [AST]) abnormalities >3xULN was ~2% per year
- most of these events were mild and did not lead to discontinuation of the drug.
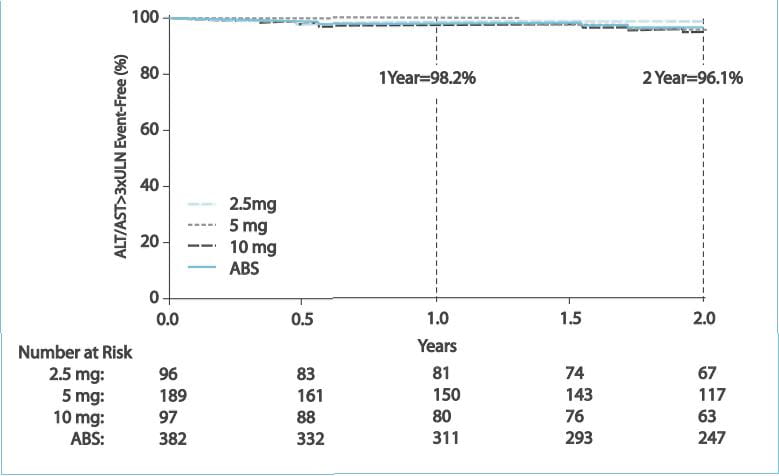
Ambrisentan treatment for 2 years was associated with sustained
improvements in the exercise capacity and a low risk of clinical worsening and death in patients with PAH.
Ambrisentan was generally well tolerated and had a low risk of aminotransferase abnormalities over the 2-year study
period.
ARIES-3 was an open-label study evaluating the efficacy and safety of ambrisentan in patients with
PAH and pulmonary hypertension due to other non-PAH aetiologies. Patients received 5 mg ambrisentan once daily for
24 weeks. The primary endpoint was the change from baseline in the 6MWD at week 24. Altogether, 224 patients were
enrolled: 62% had PAH and 38% had pulmonary hypertension due to other aetiologies. All patients had the WHO
functional class II (29%) and III (65%) symptoms at baseline, with a mean 6MWD of 317-84 metres and a median BNP
level of 199 pg/mL. At baseline, 52% were receiving sildenafil and/or prostanoid therapy. The change from the
baseline 6MWD at 24 weeks was +20.6 metres. The BNP decreased 25%, and significant improvements in the Borg dyspnoea
index and the WHO functional class were also observed. Frequent adverse events were peripheral oedema (31%),
headache (25%) and dyspnoea (15%). Aminotransferase concentrations >3xULN were reported in 6 (2.7%) patients
during the 24-week period. Thus, it was concluded that ambrisentan was well tolerated and demonstrated clinical
benefits in a broad population of patients with various pulmonary hypertension aetiologies and background pulmonary
hypertension medications.
Some ETRAs are associated with liver function test result
abnormalities. However, ambrisentan has an incidence of serum aminotransferase levels >3 ULN, similar to that
observed in PAH patients who are not receiving ETRAs. Because ambrisentan may provide benefits in PAH patients who
have discontinued ETRA therapy due to liver function test abnormalities, the safety and efficacy of ambrisentan in
this patient population was evaluated.
Inclusion Criteria
- Patients (12 to 75 years of age) with IPAH, FPAH, or APAH with connective tissue disease, congenital
systemic-to-pulmonary shunts, anorexigen use or HIV infection, and who had previously discontinued bosentan
therapy, or both, due to serum ALT and/or AST concentrations >3 ULN (liver function test result
abnormalities) were eligible for this study.
- Patients were required to have normal (<1 ULN) serum aminotransferase concentrations and a 6MWD 150
metres.
- Patients receiving sildenafil and/or a prostanoid (epoprostenol, treprostinil, iloprost) were required to
have been receiving stable therapy for 4 weeks prior to screening.
- Female patients were required to have a negative pregnancy test result, and to use a double method of
contraception during and for at least 4 weeks following their participation.
Exclusion Criteria
- Patients with pulmonary hypertension due to coronary artery disease, left-heart disease, interstitial lung
disease, chronic obstructive pulmonary disease (COPD), veno-occlusive disease, chronic thrombotic and/or
embolic disease or sleep apnoea, and portopulmonary hypertension.
- Patients having a total lung capacity <70% of the predicted normal or a forced expiratory volume in 1
second (FEV1) <65% of the predicted normal; a haemoglobin concentration<10 g/dL or
haematocrit <30%; or a resting arterial oxygen saturation <90% and refractory to treatment with oxygen
supplementation.
Study Design
- Patients who previously discontinued bosentan due to liver function test abnormalities received ambrisentan,
2.5 mg q.d. For 4 weeks, followed by 5 mg/day for 8 weeks.
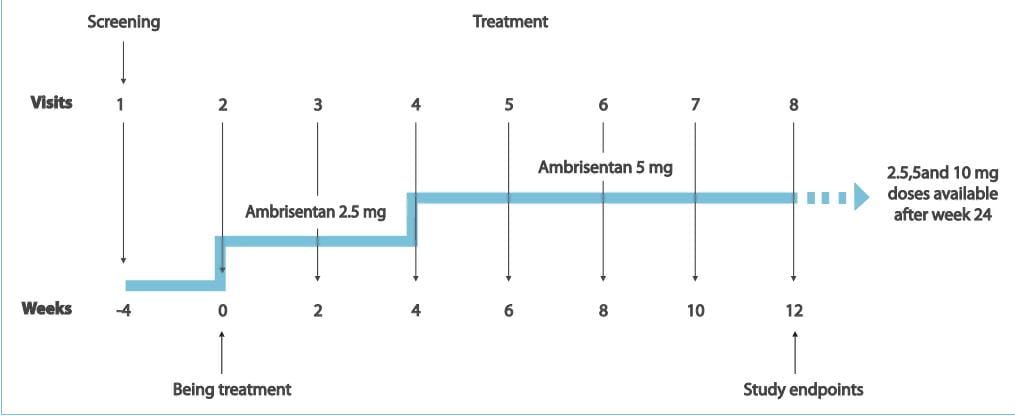
The primary endpoint was the incidence of aminotransferase levels >3xULN. Secondary end points
included the following:
- Aminotransferase levels >5 ULN requiring drug discontinuation and >3 ULN requiring dose reduction 2.
Changes in the 6MWD 3. Borg dyspnoea index 4. WHO functional class 5. Short Form-36 health survey score
Patients continued treatment beyond the 12-week endpoint with monthly monitoring of liver function
tests.
Results
- No patient had an aminotransferase level >3 ULN that required ambrisentan discontinuation. 2. In 1
patient, there was a transient aminotransferase level >3 ULN that resolved following a temporary dose
reduction. No additional aminotransferase levels >3 ULN were observed with long-term treatment (median
exposure, 102 weeks), despite dose increases to 10 mg q.d. In more than half of the patients. 3. Significant
improvement was seen in the overall survival, 6MWD, Borg dyspnoea index scale and the WHO functional class.
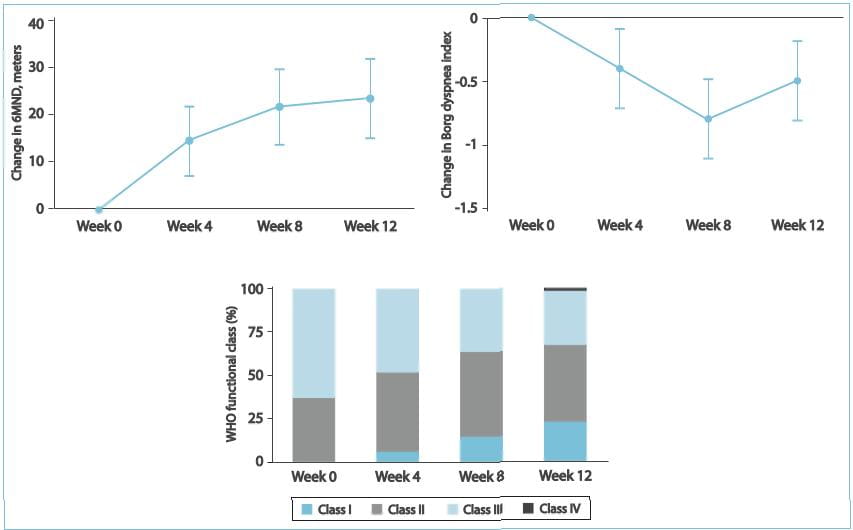
Therefore, it was concluded by this study that ambrisentan treatment may be an option for patients
who have discontinued bosentan therapy due to liver function test result abnormalities.
Ambrisentan is indicated for the treatment of PAH (Group1) in patients with the WHO functional
class II or III symptoms in order to improve the exercise capacity and delay clinical worsening
Initiate treatment at 5 mg once daily with or without food, and consider increasing the dose to 10 mg once daily if 5
mg is tolerated. Tablets may be administered with or without food. Tablets should not be split, crushed or
chewed.Doses higher than 10 mg once daily have not been studied in patients with PAH.
Ambrisentan is not recommended for use in patients <18 years of age due to a lack of data on
safety and efficacy.
No dose adjustment is required in patients aged 65 years.
Treat women of childbearing potential only after a negative pregnancy test and treat only women who are
using two acceptable methods of contraception unless the patient has had a tubal sterilization or chooses to use
a Copper T-380A IUD or LNg 20 IUS, in which case, no additional contraception is needed. Pregnancy tests should
be obtained monthly in women of childbearing potential taking ambrisentan.
Ambrisentan is not recommended in patients with moderate or severe hepatic impairment.
There is no information on the use of ambrisentan in patients with mild hepatic impairment; however, since the
main routes of metabolism of ambrisentan are glucuronidation and oxidation with subsequent elimination in the
bile, hepatic impairment would be expected to increase exposure (Cmax and AUC) to ambrisentan. Therefore,
ambrisentan should not be initiated in patients with severe hepatic impairment or clinically significant
elevated hepatic aminotransferases (>3xULN).
No dose adjustment is required in patients with renal impairment. There is limited experience with ambrisentan in
individuals with severe renal impairment (creatinine clearance <30 mL/min)
Initiate therapy cautiously in this subgroup and take particular care if the dose is increased to 10 mg.
Ambrisentan is contraindicated in patients with a history of hypersensitivity to the active
substance, to soya or to any of the excipients.
Ambrisentan may cause foetal harm when administered to a pregnant woman. Ambrisentan was teratogenic
at oral doses of 15 mg/kg/day in rats and 7 mg/kg/day in rabbits; it was not studied at lower doses. In both
species, there were abnormalities of the lower jaw and hard and soft palate, malformation of the heart and great
vessels, and failure of formation of the thymus and thyroid. Teratogenicity is a class effect of ETRAs. There are no
data on the use of ambrisentan in pregnant women. Ambrisentan is contraindicated in women who are or may become
pregnant. If this drug is used during pregnancy or if the patient becomes pregnant while taking this drug, the
patient should be apprised of the potential hazard to a foetus. Pregnancy must be excluded before the initiation of
treatment with ambrisentan and prevented during treatment and for 1 month after stopping treatment by the use of two
acceptable methods of contraception. If the patient has had a tubal sterilization or chooses to use a Copper T-380A
IUD or LNg 20 IUS for pregnancy prevention, no additional contraception is needed. Ambrisentan is contraindicated in
women of childbearing potential who are not using reliable contraception, and during lactation.
Ambrisentan has not been studied in a sufficient number of patients to establish the benefit/risk
balance in the WHO functional class IPAH. The efficacy of ambrisentan as monotherapy has not been established in
patients with the WHO functional class IV PAH. Therapy that is recommended at the severe stage of the disease (e.g.,
epoprostenol) should be considered if the clinical condition deteriorates.
Reductions in haemoglobin concentrations and haematocrit have been associated with ETRAs, including
ambrisentan. Most of these decreases were detected during the first few weeks of treatment and the haemoglobin level
generally stabilized thereafter. Marked decreases in haemoglobin (>15% decrease from baseline, resulting in a
value below the lower limit of normal) were observed in 7% of all patients receiving ambrisentan (and 10% of
patients receiving 10 mg), compared to 4% of patients receiving placebo. The cause of the decrease in haemoglobin is
unknown, but it does not appear to result from haemorrhage or haemolysis. Therefore, the haemoglobin should be
measured prior to the initiation of ambrisentan, at 1 month and periodically thereafter. Initiation of ambrisentan
therapy is not recommended for patients with clinically significant anaemia. If a clinically significant decrease in
haemoglobin is observed and other causes have been excluded, consider discontinuing ambrisentan.
Peripheral oedema has been observed with ETRAs, including ambrisentan. Most cases of peripheral
oedema in clinical studies with ambrisentan were mild to moderate in severity, although it appeared to occur with
greater frequency and severity in patients aged 65 years. Peripheral oedema was reported more frequently with 10 mg
ambrisentan. Postmarketing reports of fluid retention occurring within weeks after starting ambrisentan have been
received and, in some cases, have required intervention with a diuretic or hospitalization for fluid management or
decompensated heart failure. If patients have pre-existing fluid overload, this should be managed as clinically
appropriate prior to starting ambrisentan. If clinically significant fluid retention develops during therapy with
ambrisentan, with or without associated weight gain, further evaluation should be undertaken to determine the cause,
such as ambrisentan or underlying heart failure, and the possible need for specific treatment or discontinuation of
ambrisentan therapy.
In a 6-month study of another ETRA, bosentan, 25 male patients with the WHO functional class III and
IV PAH and normal baseline sperm count were evaluated for effects on testicular function. There was a decline of at
least 50% in the sperm count in 25% of the patients after 3 or 6 months of treatment with bosentan. One patient
developed marked oligospermia at 3 months and the sperm count remained low with two follow-up measurements over the
subsequent 6 weeks. Bosentan was discontinued and after 2 months, the sperm count had returned to baseline levels.
In 22 patients who completed 6 months of treatment, the sperm count remained within the normal range and no changes
in sperm morphology, sperm motility or hormone levels were observed. Based on these findings and preclinical data
from ETRAs, it cannot be excluded that ETRAs such as ambrisentan have an adverse effect on spermatogenesis.
If patients develop acute pulmonary oedema during initiation of therapy with vasodilating agents such
as ambrisentan, the possibility of pulmonary veno-occlusive disease should be considered and, if confirmed,
ambrisentan should be discontinued.
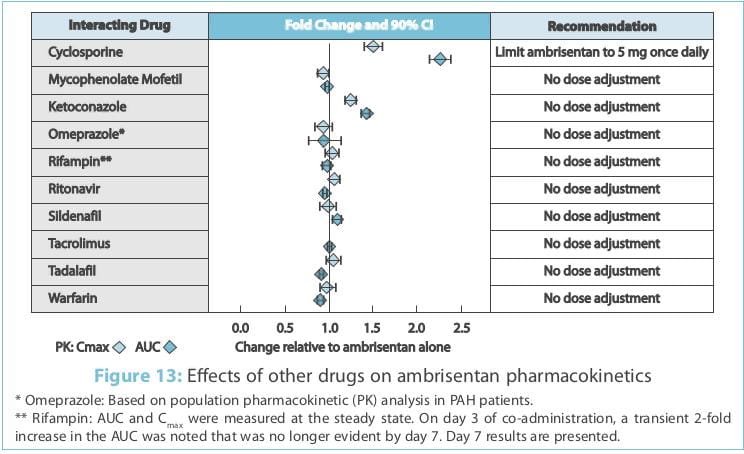
Multiple-dose co-administration of ambrisentan and cyclosporine resulted in an approximately 2-fold
increase in ambrisentan exposure in healthy volunteers; therefore, limit the dose of ambrisentan to 5 mg once daily
when co-administered with cyclosporine.
Studies with human liver tissue indicate that ambrisentan is metabolized by CYP3A, CYP2C19 and
uridine 5 -diphosphate glucuronosyltransferases ([UGTs]; 1A9S, 2B7S and 1A3S). In vitro studies suggest
that ambrisentan is a substrate of organic anion transporting polypeptides (OATP1B1 and OATP1B3), and a substrate
but not an inhibitor of P-glycoprotein (P-gp). Drug interactions might be expected because of these factors;
however, a clinically relevant interaction has been demonstrated only with cyclosporine. Ambrisentan does not
inhibit or induce drug-metabolizing enzymes at clinically relevant concentrations.
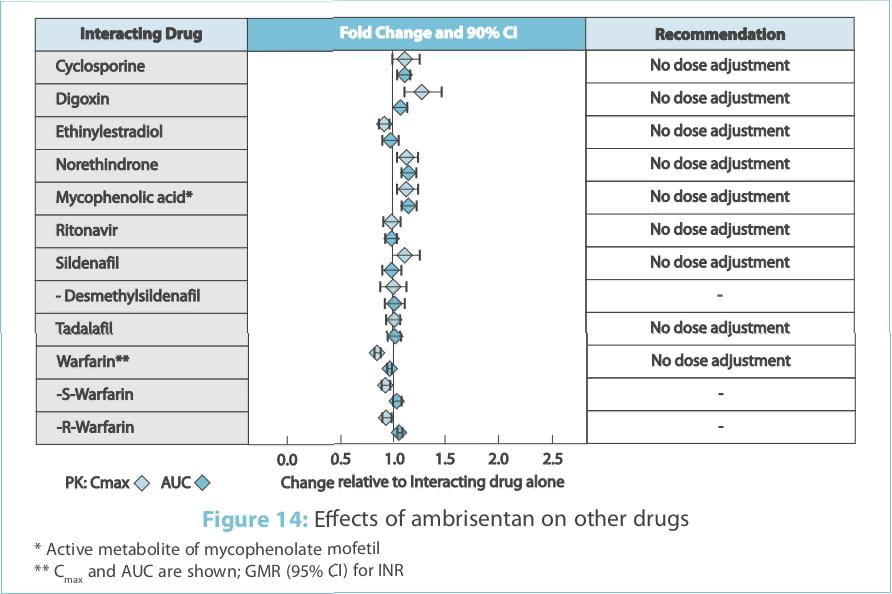
The effects of other drugs on the ambrisentan pharmacokinetics and the effects of ambrisentan on the
exposure to other drugs are shown in Figure 13 and Figure 14, respectively.
The impact of renal impairment on the pharmacokinetics of ambrisentan has been examined using a
population pharmacokinetic approach in PAH patients with creatinine clearances ranging between 20 and 150 mL/min.
There was no significant impact of mild or moderate renal impairment on exposure to ambrisentan. Dose adjustment of
ambrisentan in patients with mild or moderate renal impairment is, therefore, not required. There is no information
on the exposure to ambrisentan in patients with severe renal impairment. The impact of haemodialysis on the
disposition of ambrisentan has not been investigated.
The influence of pre-existing hepatic impairment on the pharmacokinetics of ambrisentan has not been
evaluated. Because there is in vitro and in vivo evidence of significant metabolic and biliary
contribution to the elimination of ambrisentan, hepatic impairment would be expected to have significant effects on
the pharmacokinetics of ambrisentan. Ambrisentan is not recommended in patients with moderate or severe hepatic
impairment. There is no information on the use of ambrisentan in patients with mild, pre-existing impaired liver
function; however, exposure to ambrisentan may be increased in these patients.
Pregnancy Category X
Ambrisentan is contraindicated in pregnancy. Animal studies have shown that ambrisentan is teratogenic. There is no
experience in humans. Ambrisentan treatment must not be initiated in women of childbearing potential unless the
result of a pre-treatment pregnancy test is negative and reliable contraception is practiced. Monthly pregnancy
tests during treatment with ambrisentan are recommended. Women receiving ambrisentan must be advised of the risk of
foetal harm and alternative therapy initiated if pregnancy occurs.
It is not known whether ambrisentan is excreted in human milk. Breastfeeding while receiving
ambrisentan is not recommended. A preclinical study in rats has shown decreased survival of newborn pups (mid and
high doses) and effects on the testicle size and fertility of the pups (high dose) following maternal treatment with
ambrisentan from late gestation through weaning. Doses tested were 17x51x and 170x (low, mid and high dose,
respectively) the maximum oral human dose of 10 mg on a mg/mm2 basis.
Safety and effectiveness of ambrisentan in paediatric patients have not been established.
In two placebo-controlled clinical studies of ambrisentan, 21% of the patients were 65 years old and
5% were 75 years old. The elderly (age 65 years) showed less improvement in walk distances with ambrisentan than
younger patients did, but the results of such subgroup analyses must be interpreted cautiously. Peripheral oedema
was more common in the elderly than in younger patients.
Ambrisentan was generally well tolerated, with most adverse events being mild to moderate in
intensity for all treatment groups. Peripheral oedema, headache and nasal congestion tended to be more frequent in
patients treated with ambrisentan compared with placebo. In the ARIES studies 1 and 2, none of the 261 patients
receiving ambrisentan developed serum aminotransferase concentrations >3xULN compared with 3 patients (2.3%) in
the placebo groups. Moreover, the mean values for ALT, AST, total bilirubin and alkaline phosphatase did not
increase from the baseline in the ambrisentan groups. In the ARIES-E study, the most common adverse events
encountered during the 2-year treatment period were peripheral oedema, headache, upper respiratory tract infection
and dizziness. Patients who had discontinued bosentan due to elevated liver enzymes and were switched to ambrisentan
showed no liver function test abnormalities. No patient had an aminotransferase level >3xULN that required
ambrisentan discontinuation. No additional aminotransferase levels >3xULN were observed with long-term treatment
(median exposure, 102 weeks), despite dose increases to 10 mg q.d. In more than half of the patients.
The following could be the possible explanation for decreased liver toxicity observed with
ambrisentan:
- Ambrisentan is a propanoic acid derivative whereas bosentan is a sulphonamide-based structure. This structural
variance confers differing off-target binding affinities to the hepatic transporters. Therefore, ambrisentan has
a lesser uptake in the liver cells than compared to bosentan and, thus, reduced liver toxicity.
- Bosentan, but not ambrisentan, inhibits human hepatic transporters, which provides a potential mechanism for the
increased hepatotoxicity observed for these agents in a clinical setting.
The diagnosis and management of PAH has evolved substantially in the past decade, before which it
was considered to be untreatable and fatal. Earlier diagnosis, advanced understanding of the pathogenic and
molecular pathways and a growing armamentarium of drugs have all assisted in changing the course of this challenging
disease. The guidelines recommend that all patients with PAH undergo acute vasoreactivity testing to evaluate the
response to vasodilators. For patients who show a favourable response, treatment with calcium channel blockers
should be initiated. However, if the response is inadequate, treatment with other vasodilators (prostacyclin
analogues, ETRAs or PDE-5 inhibitors) is recommended for patients with New York Heart Association (NYHA) class II,
III or IV symptoms. Patients who do not respond to initial monotherapy or who initially benefit but then deteriorate
on a single agent should be initiated with a combination of vasodilators. Ambrisentan, the selective ETRA, received
approval in 2007 from the US FDA. The approved indication was for the use in PAH (group 1) patients with the WHO
functional class II or III symptoms. It may be especially useful in patients with IPAH and PAH associated with
connective tissue disease, scleroderma, HIV and Eisenmenger's syndrome. Data available from the ARIES-E study
suggest that ambrisentan can be safely used as a long-term therapy in PAH patients. Also, with the recent safety
data available, ambrisentan can be used in PAH patients who have discontinued bosentan due to liver function test
abnormalities.
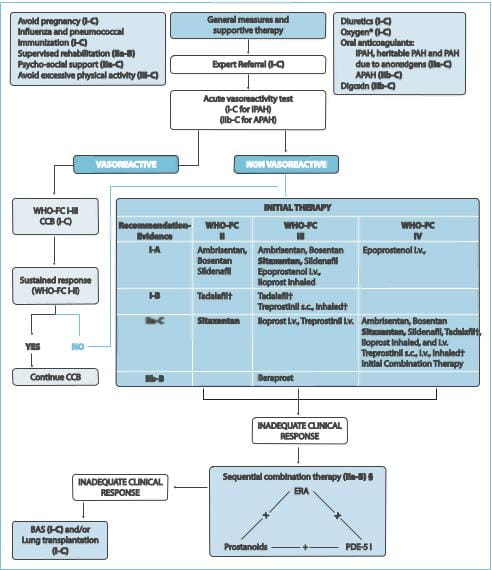
CCB=calcium channel blockers; FC=functional class; BAS=
For the use of a Registered Cardiologist only Ambrisentan Tablets
Ambrisentan is very likely to produce serious birth defects if used by pregnant women, as this
effect has been seen consistently when it is administered to animals. Pregnancy must, therefore, be excluded before
the initiation of treatment with ambrisentan, and prevented during treatment and for 1 month after stopping
treatment by the use of two acceptable methods of contraception unless the patient has had a tubal sterilization or
chooses to use a Copper T-380A IUD or LNg 20 IUS, in which case no additional contraception is needed. Monthly
pregnancy tests should also be obtained.
Endobloc 5 Tablets
Each tablet contains: Ambrisentan ... 5 mg
Endobloc 10 Tablets
Each tablet contains: Ambrisentan... 10 mg
Tablet
Pharmacodynamics
Ambrisentan is an orally active, propanoic acid-class, endothelin-receptor antagonist (ETRA), selective for the
endothelin-A (ETA)-receptor. ET plays a significant role in the pathophysiology of pulmonary arterial
hypertension (PAH). Ambrisentan is a potent (Ki 0.016 nM) and highly selective ETA-antagonist
(approximately 4,000-fold more selective for ETA as compared to ETB). Ambrisentan blocks the
ETA receptor subtype, localized predominantly on vascular smooth muscle cells and cardiac myocytes. This prevents
ET-mediated activation of second messenger systems that result in vasoconstriction and smooth muscle cell
proliferation. The selectivity of ambrisentan for the ETA-receptor over the ETB- receptor is
expected to retain ETB-receptor-mediated production of the vasodilators, nitric oxide and prostacyclin.
In a randomized, positive- and placebo-controlled, parallel-group study, healthy subjects received either
ambrisentan 10 mg daily followed by a single dose of 40 mg, placebo followed by a single dose of moxifloxacin 400 mg
or placebo alone. Ambrisentan 10 mg daily had no significant effect on the Qtc interval. The 40 mg dose of
ambrisentan increased the mean Qtc at the tmax by 5 ms with an upper 95% confidence limit (95% CI) of 9
ms. In patients receiving ambrisentan 5-10 mg daily and not taking metabolic inhibitors, no significant QT
prolongation is expected.
Pharmacokinetics
Absorption Ambrisentan is absorbed rapidly in humans. After oral administration, maximum plasma
concentrations (Cmax) of ambrisentan typically occur around 1.5 hours post-dose under both fasted and fed
conditions. The Cmax and the area under the plasma concentration-time curve (AUC) increase
dose-proportionally over the therapeutic dose range. Steady state is generally achieved following 4 days of repeat
dosing. A food-effect study involving the administration of ambrisentan to healthy volunteers under fasting
conditions and with a high-fat meal indicated that the Cmax was decreased by 12% while the AUC remained
unchanged. This decrease in peak concentration is not clinically significant and, therefore, ambrisentan can be
taken with or without food. Distribution Ambrisentan is highly plasma protein-bound. The in vitro plasma
protein-binding of ambrisentan was, on average, 98.8% and independent of concentration over the range of 0.2 to 20
mcg/ml. Ambrisentan is primarily bound to albumin (96.5%) and to a lesser extent to alpha1-acid glycoprotein. The
distribution of ambrisentan into red blood cells is low, with a mean blood to plasma ratio of 0.57 and 0.61 in males
and females, respectively. Metabolism Ambrisentan is a non-sulphonamide (propanoic acid) ETRA. Ambrisentan is
glucuronidated via several uridine 5'-diphosphate glucuronosyltransferase (UGT) isoenzymes (UGT1A9S, UGT2B7S and
UGT1A3S) to form ambrisentan glucuronide (13%). Ambrisentan also undergoes oxidative metabolism mainly by cytochrome
(CY) P3A4 and to a lesser extent by CYP3A5 and CYP2C19 to form 4-hydroxymethyl ambrisentan (21%), which is further
glucuronidated to 4-hydroxymethyl ambrisentan glucuronide (5%). The binding affinity of 4-hydroxymethyl ambrisentan
for the human ET-receptor is 65-fold less than ambrisentan. Therefore, at concentrations observed in the plasma
(approximately 4% relative to the parent ambrisentan), 4-hydroxymethyl ambrisentan is not expected to contribute to
the pharmacological activity of ambrisentan. In vitro data have shown that at therapeutic concentrations,
ambrisentan does not inhibit UGT1A1, UGT1A6, UGT1A9, UGT2B7 or the CYP450 enzymes, 1A2, 2A6, 2B6, 2C8, 2C9, 2C19,
2D6, 2E1 and 3A4. Additional in vitro studies showed that ambrisentan does not inhibit NTCP, organic anion
transporting polypeptides (OATP) or BSEP. Furthermore, ambrisentan does not induce MRP2, P-glycoprotein (P-gp) or
BSEP. Elimination Ambrisentan and its metabolites are eliminated primarily in the bile following hepatic and/or
extra- hepatic metabolism. Approximately 22% of the administered dose is recovered in the urine following oral
administration, with 3.3% being unchanged ambrisentan. Plasma elimination half-life in humans ranges from 13.6 to
16.5 hours.
Ambrisentan is indicated for the treatment of PAH (World Health Organization [WHO] Group I) in
patients with the WHO functional class II or III symptoms in order to improve exercise capacity and delay clinical
worsening.
Adults
Initiate treatment at 5 mg once daily with or without food, and consider increasing the dose to 10 mg once daily
if 5 mg is tolerated. Tablets may be administered with or without food. Tablets should not be split, crushed or
chewed. Doses higher than 10 mg once daily have not been studied in patients with PAH.
Adolescents and Children
Ambrisentan is not recommended for use in patients <18 years of age due to a lack of data on
safety and efficacy.
Geriatric Use
No dose adjustment is required in patients aged 65 years.
Women of Childbearing Potential
Treat women of childbearing potential only after a negative pregnancy test and treat only women who are using two
acceptable methods of contraception unless the patient has had a tubal sterilization or chooses to use a Copper
T-380A IUD or LNg 20 IUS, in which case, no additional contraception is needed. Pregnancy tests should be obtained
monthly in women of childbearing potential taking ambrisentan.
Pre-Existing Hepatic Impairment
Ambrisentan is not recommended in patients with moderate or severe hepatic impairment. There is no
information on the use of ambrisentan in patients with mild hepatic impairment; however, since the main routes of
metabolism of ambrisentan are glucuronidation and oxidation with subsequent elimination in the bile, hepatic
impairment would be expected to increase exposure (Cmax and AUC) to ambrisentan. Therefore ambrisentan should not be
initiated in patients with severe hepatic impairment or clinically signifi cant elevated hepatic aminotransferases
(greater than 3 times the upper limit of normal [>3xULN]).
Pre-Existing Renal Impairment
No dose adjustment is required in patients with renal impairment. There is limited experience with ambrisentan in
individuals with severe renal impairment (creatinine clearance <30 mL/min)
- Initiate therapy cautiously in this subgroup and take particular care if the dose is increased to 10 mg.
Ambrisentan is contraindicated in patients with a history of hypersensitivity to the active
substance, to soya or to any of the excipients.
Ambrisentan may cause foetal harm when administered to a pregnant woman. Ambrisentan was
teratogenic at oral doses of 15 mg/kg/day in rats and 7 mg/kg/day in rabbits; it was not studied at lower doses. In
both species, there were abnormalities of the lower jaw and hard and soft palate, malformation of the heart and
great vessels, and failure of formation of the thymus and thyroid. Teratogenicity is a class effect of ETRAs. There
are no data on the use of ambrisentan in pregnant women. Ambrisentan is contraindicated in women who are or may
become pregnant. If this drug is used during pregnancy or if the patient becomes pregnant while taking this drug,
the patient should be apprised of the potential hazard to a foetus. Pregnancy must be excluded before the initiation
of treatment with ambrisentan and prevented during treatment and for 1 month after stopping treatment by the use of
two acceptable methods of contraception. If the patient has had a tubal sterilization or chooses to use a Copper
T-380A IUD or LNg 20 IUS for pregnancy prevention, no additional contraception is needed. Ambrisentan is
contraindicated in women of childbearing potential who are not using reliable contraception, and lactation.
Ambrisentan has not been studied in a sufficient number of patients to establish the benefit/risk
balance in the WHO functional class I PAH. The efficacy of ambrisentan as monotherapy has not been established in
patients with the WHO functional class IV PAH. Therapy that is recommended at the severe stage of the disease (e.g.,
epoprostenol) should be considered if the clinical condition deteriorates.
Haematological Changes
Reductions in haemoglobin concentrations and haematocrit have been associated with ETRAs, including
ambrisentan. Most of these decreases were detected during the first few weeks of treatment and the haemoglobin level
generally stabilized thereafter. Marked decreases in haemoglobin (>15% decrease from baseline, resulting in a
value below the lower limit of normal) were observed in 7% of all patients receiving ambrisentan (and 10% of
patients receiving 10 mg), compared to 4% of patients receiving placebo. The cause of the decrease in haemoglobin is
unknown, but it does not appear to result from haemorrhage or haemolysis. Therefore, the haemoglobin should be
measured prior to the initiation of ambrisentan, at 1 month and periodically thereafter. Initiation of ambrisentan
therapy is not recommended for patients with clinically significant anaemia. If a clinically significant decrease in
haemoglobin is observed and other causes have been excluded, consider discontinuing ambrisentan.
Fluid Retention
Peripheral oedema has been observed with ETRAs, including ambrisentan. Most cases of peripheral
oedema in clinical studies with ambrisentan were mild to moderate in severity, although it appeared to occur with
greater frequency and severity in patients aged 65 years. Peripheral oedema was reported more frequently with 10 mg
ambrisentan. Postmarketing reports of fluid retention occurring within weeks after starting ambrisentan have been
received and, in some cases, have required intervention with a diuretic or hospitalization for fluid management or
decompensated heart failure. If patients have pre-existing fluid overload, this should be managed as clinically
appropriate prior to starting ambrisentan. If clinically significant fluid retention develops during therapy with
ambrisentan, with or without associated weight gain, further evaluation should be undertaken to determine the cause,
such as ambrisentan or underlying heart failure, and the possible need for specific treatment or discontinuation of
ambrisentan therapy.
Decreased Sperm Counts
In a 6-month study of another ETRA, bosentan, 25 male patients with the WHO functional class III
and IV PAH and normal baseline sperm count were evaluated for eff ects on testicular function. There was a decline
of at least 50% in the sperm count in 25% of the patients after 3 or 6 months of treatment with bosentan. One
patient developed marked oligospermia at 3 months and the sperm count remained low with two follow-up measurements
over the subsequent 6 weeks. Bosentan was discontinued and after 2 months, the sperm count had returned to baseline
levels. In 22 patients who completed 6 months of treatment, the sperm count remained within the normal range and no
changes in sperm morphology, sperm motility or hormone levels were observed. Based on these fi ndings and
preclinical data from ETRAs, it cannot be excluded that ETRAs such as ambrisentan have an adverse eff ect on
spermatogenesis.
Pulmonary Veno-Occlusive Disease
If patients develop acute pulmonary oedema during initiation of therapy with vasodilating agents
such as ambrisentan, the possibility of pulmonary veno-occlusive disease should be considered and, if confi rmed,
ambrisentan should be discontinued.
Multiple-dose co-administration of ambrisentan and cyclosporine resulted in an approximately 2-fold
increase in ambrisentan exposure in healthy volunteers; therefore, limit the dose of ambrisentan to 5 mg once daily
when co-administered with cyclosporine.
In Vitro Studies
Studies with human liver tissue indicate that ambrisentan is metabolized by CYP3A, CYP2C19 and UGTs
(1A9S, 2B7S and 1A3S). In vitro studies suggest that ambrisentan is a substrate of OATP1B1 and OATP1B3, and
a substrate but not an inhibitor of P-gp. Drug interactions might be expected because of these factors; however, a
clinically relevant interaction has been demonstrated only with cyclosporine. Ambrisentan does not inhibit or induce
drug-metabolizing enzymes at clinically relevant concentrations.
In Vivo Studies
The effects of other drugs on the ambrisentan pharmacokinetics and the eff ects of ambrisentan on
the exposure to other drugs are shown in Figure 1 and Figure 2, respectively.
On day 3 of co-administration, a transient 2-fold increase in the AUC was noted that was no longer
evident by day 7. Day 7 results are presented.
Renal Impairment
The impact of renal impairment on the pharmacokinetics of ambrisentan has been examined using a population
pharmacokinetic approach in PAH patients with creatinine clearances ranging between 20 and 150 mL/min. There was no
significant impact of mild or moderate renal impairment on exposure to ambrisentan. Dose adjustment of ambrisentan
in patients with mild or moderate renal impairment is, therefore, not required. There is no information on the
exposure to ambrisentan in patients with severe renal impairment. The impact of haemodialysis on the disposition of
ambrisentan has not been investigated.
Hepatic Impairment
The influence of pre-existing hepatic impairment on the pharmacokinetics of ambrisentan has not
been evaluated. Because there is in vitro and in vivo evidence of significant metabolic and
biliary contribution to the elimination of ambrisentan, hepatic impairment would be expected to have significant
effects on the pharmacokinetics of ambrisentan. Ambrisentan is not recommended in patients with moderate or severe
hepatic impairment. There is no information on the use of ambrisentan in patients with mild, pre-existing impaired
liver function; however, exposure to ambrisentan may be increased in these patients.
Pregnancy
Pregnancy Category X
Ambrisentan is contraindicated in pregnancy. Animal studies have shown that ambrisentan is teratogenic. There is no
experience in humans. Ambrisentan treatment must not be initiated in women of childbearing potential unless the
result of a pre-treatment pregnancy test is negative and reliable contraception is practiced. Monthly pregnancy
tests during treatment with ambrisentan are recommended. Women receiving ambrisentan must be advised of the risk of
foetal harm and alternative therapy initiated if pregnancy occurs.
Lactation
It is not known whether ambrisentan is excreted in human milk. Breastfeeding while receiving
ambrisentan is not recommended. A preclinical study in rats has shown decreased survival of newborn pups (mid and
high doses) and effects on the testicle size and fertility of the pups (high dose) following maternal treatment with
ambrisentan from late gestation through weaning. Doses tested were 17x51x and 170x (low, mid and high dose,
respectively) the maximum oral human dose of 10 mg on a mg/mm2 basis.
Paediatric Use
Safety and effectiveness of ambrisentan in paediatric patients have not been established.
Geriatric Use
In two placebo-controlled clinical studies of ambrisentan, 21% of the patients were 65 years old
and 5% were 75 years old. The elderly (age 65 years) showed less improvement in walk distances with ambrisentan than
younger patients did, but the results of such subgroup analyses must be interpreted cautiously. Peripheral oedema
was more common in the elderly than in younger patients.
Experience from Clinical Studies
The safety of ambrisentan has been evaluated in clinical trials of more than 483 patients with PAH. Adverse drug
reactions identified from 12-week, placebo-controlled, clinical trial data are listed below by system organ class
and frequency. With longer observation in uncontrolled studies (mean observation of 79 weeks), the safety profile
was similar to that observed in the short-term studies. Frequencies were defined as follows: very common (1/10);
common (1/100 to <1/10); uncommon (1/1,000 to <1/100); rare (1/10,000 to <1/1,000); and, very rare
(<1/10,000). For dose-related adverse reactions, the frequency category reflects the higher dose of ambrisentan.
Frequency categories do not account for other factors, including varying study duration, pre-existing conditions and
baseline patient characteristics. Adverse reaction frequency categories assigned (based on clinical trial
experience) may not reflect the frequency of adverse events occurring during normal clinical practice. Within each
frequency grouping, undesirable effects are presented in order of decreasing seriousness.
Cardiac Disorders
Common: Palpitation.
Blood and Lymphatic System Disorders
Common: Anaemia (decreased haemoglobin, decreased haematocrit).
Nervous System Disorders
Very common: Headache (including sinus headache, migraine1).
Respiratory, Thoracic and Mediastinal
Disorders
Common: Upper respiratory (e.g., nasal,2 sinus) congestion, sinusitis, nasopharyngitis,
rhinitis.
Gastrointestinal Disorders
Common: Abdominal pain, constipation.
Vascular Disorders
Common: Flushing.
General Disorders and Administration Site
Conditions
Very common: Peripheral oedema, fluid retention.3
Common: Chest pain/discomfort.
Immune System Disorders
Uncommon: Hypersensitivity reactions (e.g., angio-oedema, rash, pruritus).
1 The frequency of headache appeared higher with 10 mg ambrisentan.
2 The incidence of nasal congestion was dose-related during ambrisentan therapy.
3 Peripheral oedema was reported more frequently with 10 mg ambrisentan.
In clinical studies, peripheral oedema was reported more commonly and tended to be more severe in patients aged
65 years.
Laboratory Abnormalities
Decreased Haemoglobin
The frequency of decreased haemoglobin (anaemia) was higher with 10 mg ambrisentan. Across the 12-week,
placebo-controlled, Phase III clinical studies, mean haemoglobin concentrations decreased for patients in the
ambrisentan groups and were detected as early as week 4 (decrease by 0.83 g/dL); mean changes from baseline appeared
to stabilize over the subsequent 8 weeks. A total of 17 patients (6.5%) in the ambrisentan treatment groups had
decreases in haemoglobin of 15% from baseline, which fell below the lower limit of normal.
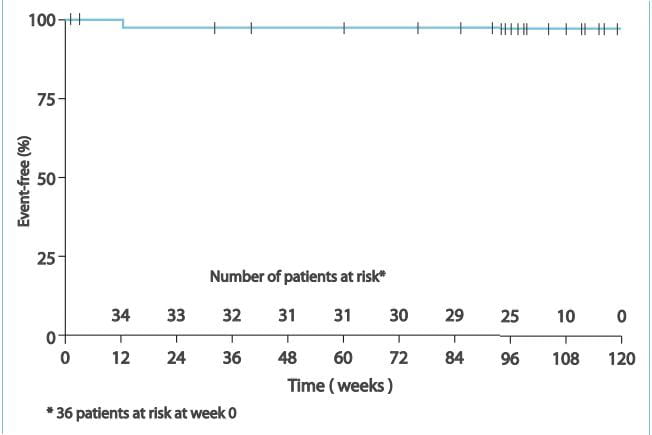
Use in Patients with Prior ETRA-Related Serum Liver Enzyme Abnormalities
In an uncontrolled, open-label study, 36 patients who had previously discontinued ETRAs due to
aminotransferase elevations >3xULN were treated with ambrisentan. All patients had to have normal
aminotransferase levels on entry to this study. Prior elevations were predominantly moderate, with 64% of the ALT
elevations <5xULN, but 9 patients had elevations >8xULN. In 8 patients who had been rechallenged with bosentan
and/or the investigational ETRA, all 8 had a recurrence of aminotransferase abnormalities that required
discontinuation of ETRA therapy. Of the 36 patients, 25 were also receiving prostanoid and/or phosphodiesterase type
5 (PDE-5) inhibitor therapy. There was early discontinuation by 2 patients (including one of the patients with a
prior 8xULN elevation). Of the remaining 34 patients, 1 patient experienced a mild aminotransferase elevation at 12
weeks on ambrisentan 5 mg, which was resolved by decreasing the dosage to 2.5 mg; this did not recur with later
escalations to 10 mg. With a median follow-up of 13 months and with 50% of patients increasing the dose of
ambrisentan to 10 mg, no patients were discontinued for aminotransferase elevations. While the uncontrolled study
design does not provide information about what would have occurred with re-administration of previously used ETRAs
or show that ambrisentan led to fewer aminotransferase elevations than would have been seen with those drugs, the
study indicates that ambrisentan may be tried in patients who have experienced asymptomatic aminotransferase
elevations on other ETRAs after aminotransferase levels have returned to normal.
Postmarketing Data
In addition to adverse reactions identified from clinical studies, the following adverse reactions
were identified during post-approval use of ambrisentan. Frequencies were defined as follows: common (1/100 to
<1/10); uncommon (1/1,000 to <1/100); and, not known (cannot be estimated from the available data).
Nervous System Disorders
Not known: Dizziness.
Cardiac Disorders
Not known: Cardiac failure4.
Vascular Disorders
Not known: Syncope, hypotension.
Respiratory, Thoracic and Mediastinal
Disorders
Not known: Dyspnoea5.
Gastrointestinal Disorders
Not known: Syncope, hypotension.
Hepatobiliary Disorders
Common: Hepatic transaminases increased.
Uncommon: Hepatic injury and autoimmune hepatitis6.
Immune System Disorders
Hypersensitivity reactions (e.g., angio-oedema, rash).
Others
Anaemia, nausea, and vomiting.
Elevations of liver aminotransferases (alanine aminotransferase [ALT] and/or aspartate
aminotransferase [AST]) have been reported with ambrisentan use; in most cases, alternative causes of the
liver injury could be identified (heart failure, hepatic congestion, hepatitis, alcohol use, hepatotoxic
medications). Other ETRAs have been associated with elevations of aminotransferases, hepatotoxicity and
cases of liver failure. Discontinue ambrisentan if >5xULN or if elevations are accompanied by bilirubin
>2xULN or by signs or symptoms of liver dysfunction and other causes are excluded. Because these
reactions were reported voluntarily from a population of uncertain size, it is not possible to reliably
estimate the frequency or establish a causal relationship to drug exposure.
4Most of the reported cases of cardiac failure were associated with fluid retention.
5Cases of worsening dyspnoea of unclear aetiology have been reported shortly after starting ambrisentan
therapy.
6 Cases of autoimmune hepatitis, including cases of exacerbation of autoimmune hepatitis, and
hepatic injury have been reported during ambrisentan therapy.
There is no experience with overdosage of ambrisentan. The highest single dose of ambrisentan administered to
healthy volunteers was 100 mg and the highest daily dose administered to patients with PAH was 10 mg once daily.
In healthy volunteers, single doses of 50 mg and 100 mg (5 to 10 times the maximum recommended dose) were
associated with headache, flushing, dizziness, nausea and nasal congestion. Massive overdosage could potentially
result in hypotension. In the case of pronounced hypotension, active cardiovascular support may be required. No
specific antidote is available.
1. European Heart Journal 2009; 30:2493-2537
2. CHEST 2001; 120:1562-1569
3. Am J Cardiovasc Drugs 2011; 11:215-226
4. Circulation 2008; 117:3010-3019
5. J Am Coll Cardiol 2009; 54:1971-1981
6. CHEST 2009; 135:122-129
7. Can. J. Physiol. Pharmacol. 2010; 88:682-691
8. Am J Respir Crit Care Med 179; 2009:A3357