

Antibacterial agents are classified based on chemical structure and proposed mechanism of action as follows:1
ABC of Anti-infectives : A Handbook on Anti-infective Armamentarium

27 Jun, 12


ABC of Anti-infectives : A Handbook on Anti-infective Armamentarium
Anti-Bacterials
Classification of Antibacterial Agents
- Agents that inhibit synthesis of bacterial cell wall, including beta-lactam class and dissimilar agents such as vancomcyin and bacitracin.
- Agents that act directly on the cell membrane of the microorganism, such as polymyxin; and lipopepetide daptomycin.
- Agents that disrupt the function of 30 S or 50 S ribosomal subunits to reversibly inhibit protein synthesis which generally are bacteriostatic e.g., chloramphenicol, tetracyclines, streptogramins, clindamycin and linezolid.
- Agents that bind to 30 S ribosomal subunits and alter protein synthesis, which generally are bactericidal e.g., aminoglycosides.
- Agents that aff ect bacterial nucleic acid and metabolism such as quinolones.
- The antimetabolites, including trimethoprim and sulfonamides.
Beta-Lactams
Beta -lactam antimicrobials have been widely prescribed to treat serious infections for nearly 60 years owing to their excellent efficacy, safety and tolerability profiles.2
The mechanism of action of beta-lactam antimicrobial agents (penicillins, cephalosporins, monobactams and carbapenems) is related to their ability to inhibit bacterial cell wall synthesis and in some organisms trigger lysis of the cell wall. Beta-lactams bind to various intracellular proteins called penicillin-binding proteins (PBPs) and exert their effects at several stages of cell wall synthesis. The affinity and specificity of different PBPs may help to explain why the various agents have differing spectrums of activity. Alteration in binding to certain PBPs has also been associated with antimicrobial resistance.3
Figure 1 : Classification of cell-wall inhibitors
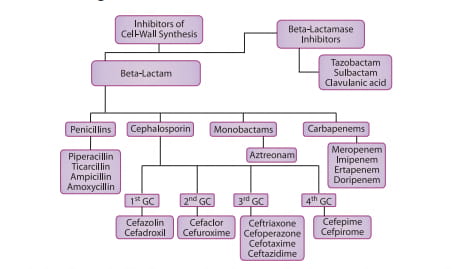
Penicillins
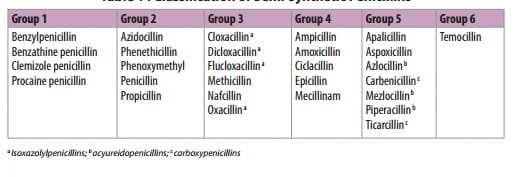
The history of the brilliant research that led to the discovery and development of penicillin is well chronicled. The basic structure of penicillins consists of a thiazolidinone ring fused with a beta-lactam ring which is essential for antibacterial activity. The two rings constitute the fundamental nucleus of all the penicillins, namely, 6 aminopenicillanic acid (6-APA). A variety of semi-synthetic penicillins (Table 1) are produced by altering the composition of the side chain attached to 6-APA nucleus. Both 6-APA nucleus and the side chain are essential for the antibacterial activity, but the side chain, in addition, determines the stability of the penicillin against degradation by gastric acid and the enzyme penicillinase (beta-lactamase) produced by certain microorganisms.4
The large number of semi synthetic penicillins in clinical use may be divided into six groups.5
Group 1: Benzylpenicillin and its long acting parenteral forms.
Group 2: Orally absorbed penicillins similar to benzylpenicillin.
Group 3: Penicillins relatively stable to staphylococcal beta-lactamase, but which have no useful activity against gram-negative bacilli. They have less potent antimicrobial activity against microorganisms that are sensitive to penicillin G, but are agents of first choice for treatment of penicillinase producing S.aurues and S.epidermidis that are not methicillin-resistant.
Group 4: Compounds with enhanced activity against certain gram-negative bacilli, including many enterobacteria and Haemophilus infl uenza, but which are inactivated by staphylococcal and many enterobacterial beta-lactamases. They include several aminopenicillins, such as ampicillin and amoxicillin, and the amidinopenicillin and mecillinam. Various esters and condensates of ampicillin and a mecillinam ester, pivmecillinam, have been developed to improve the poor oral absorption of the parent compounds to which they owe their antimicrobial activity.
Group 5: These include carboxypenicillins (and their orally absorbed esters) and acyl or acylureido derivatives of ampicillin. They are active against P. aeruginosa, Klebsiella and other certain gram-negative microorganisms. Piperacillin retains activity of ampicillin against gram-positive cocci and L. monocytogenes and is superior to ticarcillin against Pseudomonas.
Group 6: These include penicillins that are resistant to enterobacterial beta-lactamases.
Table 1 : Classification of Semi-synthetic Penicillins
Clinical Indications5
Group 1 Penicillins are indicated for streptococcal infections, including bacteraemia, empyema, severe pneumonia, endocarditis, meningitis, staphylococcal infections, gonorrhoea and gonorrhoeal endocarditis.
They are also indicated for syphilis, anthrax, actinomycosis, clostridial infections (including tetanus), and diphtheria (to prevent carrier state).
Group 2 Penicillins may be prescribed for many indications for which benzylpenicillin is suitable, but are not recommended as initial therapy for serious infections. However they are useful as continuation therapy in osteomyelitis or endocarditis after initial control of the disease by parenteral benzylpenicillin. They are not appropriate for respiratory infections, where H. influenzae is implicated and treatment of gonorrhoea, syphilis or leptospirosis and infections due to gram-negative bacilli.
Group 3 Penicillins are an important option for proven staphylococcal infections including those of bones, joints, heart valves and meninges, brain abscess and disseminated infection.
Group 4 Penicillins are indicated for upper respiratory tract infections (other than pharyngitis, which may mask glandular fever) due to streptococci, pneumococci and H. influenzae. Others include lower respiratory tract infections, urinary tract infections, infections of skin and soft tissues, gonorrhoea and for dental prophylaxis in patients at risk of endocarditis (single high dose).
Group 5 and 6 Penicillins are indicated for serious infections with susceptible organisms including lower respiratory tract, intra-abdominal, urinary tract, gynaecological infections and septicaemia.
Cephalosporins
Cephalosporium acremonium, the first source of the cephalsporins, was isolated in 1948 by Brotzu from the sea near a sewer outlet off the Sardinina coast. With the isolation of the active nucleus of cephalosporin C, 7-aminocephalosporanic acid, and with the addition of side chains, it became possible to produce semisynthetic compounds with antibacterial activity very much greater than that of parent compound.
The first generation cephalosporins - cephalothin and cefazolin, have good activity against gram-positive bacteria and relatively modest activity against gram-negative microorganisms. Most oral cavity anaerobes are sensitive, but the B. fragilis group is resistant. The second generation cephalosporins have somewhat increased activity against gram-negative microorganisms but are much less active than the third generation agents. A subset of second generation (cefoxitin, cefotetan and cefmetazole) is also active against B. fragilis group.
Third generation cephalosporins generally are less active than first-generation agents against gram-positive cocci, but they are much more active against Enterobacteriaceae. Ceftazidime and cefoperazone are also active against Pseudomonas but less active than other third generation agents against gram-positive cocci.
Fourth generation cephalosporins, such as cefepime, have an extended spectrum of activity compared to the third generation and have an increased stability from hydrolysis by plasmid and chromosomally mediated beta-lactamases. They are particularly useful for the empirical treatment of serious infections in hospitalized patients with Pseudomonas and Enterobacteriaceae infections.
It is important to remember that none of the cephalosporins have reliable activity against the following bacteria: penicillin-resistant S. pneumoniae, methicillinresistant S. aureus and S. epidermidis, coagulase-negative Staphylococci, Enterococcus, L. monocytogenes, Legionella pneumophilia, L. micdadei, C. difficle, Xanthomonas maltophilia, Campylobacter jejuni and Acinetobacter spp.6
Clinical Indications
Cephalosporins are effective as both therapeutic and prophylaxis agents.
The first generation cephalosporins are excellent agents for skin and soft tissue infections owing to S. aureus and S. pyogenes. A single dose of cefazolin just before surgery is the preferred prophylaxis for procedures in which skin flora are likely pathogens.
Second generation agents, such as cefoxitin or cefotetan, are preferred as prophylaxis in colorectal surgery. These agents should not be used as empirical treatment for meningitis or pneumonia since they have inferior activity against penicillin-resistant S. pneumoniae compared to third generation agents or ampicillin. The oral second generation cephalosporins can be used to treat respiratory tract infections, although they are suboptimal compared to oral amoxicillin.
The third generation cephalosporins, with or without aminoglycosides, have been considered to be the drugs of choice for serious infection caused by Klebsiella, Enterobacter, Proteus, Providencia, Serratia and Haemophlius spp. Cefotaxime or ceftriaxone are generally preferred for initial treatment of meningitis in nonimmunocompromised adults and children older than 3 months of age (in combination with vancomycin and ampicillin pending identification of causative agents) because of their spectrum and good penentration into CSF. However, cefotaxime has failed in the treatment of meningitis owing to resistant S. pneumoniae, thus vancomcyin should be added.
The fourth generation cephalosporins are indicated for the empirical treatment of nosocomial infections where antibiotic resistance owing to extended-spectrum betalactamases is anticipated.6
Beta-Lactamase Inhibitors
Clavulanate, sulbactam and tazobactam are the only beta-lactamase inhibitors currently available for clinical use. They irreversibly inhibit beta-lactamases via acylenzyme complex formation, which allows the intact accompanying penicillin or cephalosporin to exhibit time-dependent bacterial killing (usually through binding to PBP-1and -3).
The overall antibacterial spectrum of these drug combinations depends on the intrinsic activity of the beta-lactam as well as the characteristics of the individual inhibitor towards different beta-lactamases. As noted, methicillin-sensitive S. aureus (MSSA), H. influenzae, Moraxella catarrhalis, Bacteroides spp. and many Enterobacteriaceae spp., such as Escherichia coli, Klebsiella and Proteus spp. are generally susceptible. Streptococci, Pneumococci, Enterococci and even Pseudomonas spp. are susceptible depending on the accompanying beta-lactam.
Combinations containing piperacillin or cefoperazone off er the broadest antibacterial spectrum. When used alone, even potent cephalosporins such as ceftazidime will not provide satisfactory activity against gram-positive pathogens or anaerobes.7
Clinical Indications
Beta-lactam/beta-lactamase inhibitor combinations are particularly useful against mixed infections. Clinical experience confirms their effectiveness in the empirical treatment of respiratory, intra-abdominal, and skin and soft tissue infections. There is evidence to suggest that they are efficacious in treating patients with neutropenic fever and nosocomial infections, especially in combination with other agents. Their role in treating various multi-resistant pathogens such as Acinetobacter spp. and Stenotrophomonas maltophilia are gaining importance. Although, generally, they do not constitute reliable therapy against extended-spectrum beta-lactamase producers, their substitution in place of cephalosporins appears to reduce emergence of the latter pathogens.7
Monobactams
Aztreonam is a monocylic beta-lactam compound (a monobactam) isolated from Chromobacterium violaceum. Aztreonam is truly a narrow spectrum antibacterial and its activity differs from those of other beta-lactam antibiotics and more closely resembles that of an aminoglycoside. Aztreonam has activity against gram-negative bacteria; however it has no activity against gram-positive bacteria and anaerobic organisms. However, activity against Enterobaceriaceae is excellent, as is that against P. aerguinosa. It is also highly active in vitro against H. influenzae and Gonococci.6
Mechanism of action
It interacts with PBPs of susceptible microorganisms and induces the formation of long filamentous bacterial structures. The compound is resistant to many beta lactamases that are elaborated by most gram-negative bacteria.6
Clinical Indications and Dosage
The usual dose of aztreonam for severe infections is 2g every 6 to 8 hours. This should be reduced in patients with renal impairment.6
Carbapenems
Among the many different structurally distinct classes of beta-lactams, the carbapenem class, while sharing the general beta-lactam features, is regarded as the class that is most potent and that has the widest spectrum of antimicrobial activity.8
The carbapenems currently available in our country are imipenem (formulated with cilastatin), meropenem, ertapenem and doripenem.
Though carbapenems exhibit broad-spectrum coverage, there are some differences to a large extent determined by the variability in the side chains at the exocyclic sulfur atom. Imipenem has slightly better activity against gram-positive bacteria than does meropenem, while meropenem and ertapenem are more active against gram negative organisms than is imipenem. Meropenem may be active against P. aeruginosa isolates resistant to imipenem because meropenem enters these organisms more readily than does imipenem.9 However, ertapenem has limited in vitro activity against Pseudomonas spp. and Acinetobacter spp.8 In contrast to imipenem, meropenem and doripenem that have in vivo half lives of approximately 1 hour, ertapenem has a half life of approximately 4 hours making it suitable for once-daily administration.10
Doripenem is a promising new carbapenem with similar properties to those of meropenem, although it appears to have more potent in vitro activity against P. aeruginosa than meropenem.10
Though carbapenems are stable against most beta-lactamases,9 but (like other betalactam antibiotics) they are hydrolyzed by the intrinsically expressed metallo-betalactamase of Stenotrophomonas maltophilia and by some Bacillus and Bacteroides betalactamases. Carbapenems are inactive against some isolates of Burkholderia cepacia. Carbapenems inhibit the growth of some enterococcal species but are bacteriostatic rather than bactericidal. Furthermore, these agents lack activity against Enterococcus faecium because they do not inhibit the PBPs that this organism uses in cell wall synthesis.
Faropenem, a member of the unique penem class available as oral drug, is not a carbapenem, and thus has different chemical and microbiological properties compared to carbapenems.10 It is four- to eight-fold more active against penicillin resistant isolates of S. pneumoniae than amoxicillin/clavulanate. Faropenem also maintains its in vitro activity against multi-drug resistant strains of S. pneumoniae including strains resistant to macrolides, tetracycline, trimethoprimsulfamethoxazole and fluoroquinolones.11
Mechanism of Action
Carbapenems are bactericidal. In contrast to other beta-lactam antibiotics, and similar to aminoglycosides and fluoroquinolones, carbapenems show a significant postantibiotic eff ect against gram-negative bacterial pathogens. This post-antibiotic eff ect is most marked with P. aeruginosa and allows for less frequent dosing than would be expected based on serum levels of the drug.9
Clinical Indications and Dosage
Based on their spectrum of activity carbapenems have different clinical applications:8,10
Group 1 includes ertapenem, with limited activity against non-fermentative gram negative bacilli that is particularly suitable for community-acquired infections and as outpatient intravenous antimicrobial therapy and recommended dose is 1g once daily.
Group 2 includes imipenem, meropenem and doripenem, with activity against nonfermentative gram-negative bacilli that are particularly suitable for nosocomial infections including complicated intra-abdominal infections, skin and skin structure infections (SSSIs), community-acquired pneumonia (CAP), nosocomial pneumonia, complicated urinary tract infections, meningitis (meropenem only) and febrile neutropenia.
For the treatment of most serious infections, including ventilator-associated pneumonia (VAP), meropenem should be administered at a dosage of 1g intravenously every eight hours; if necessary; the dosage can be increased to a maximum of 2 g every eight hours,12 while imipenem/cilastatin can be dosed upto 4 gms a day.13
Doripenem is approved for a dose of 500 mg IV every six to eight hours.
Group 3 includes carbapenems with clinical activity against MRS (none currently licensed).
Aminoglycosides
Aminoglycosides are primarily used to combat infections due to aerobic and gram negative bacteria. As a group they are widely active against the Enterobacteriaceae, such as E. coli, Klebsiella spp., Enterobacter spp., Proteus spp. and Serratia spp. and other aerobic gram-negative bacilli including, for some compounds, P. aeruginosa.They are active to different degrees against S. aureus, coagulase-negative Staphylococci and Corynebacterium spp., but the activity against many other gram-positive bacteria, including Streptococci, is generally limited. However, they interact synthergistically with antibiotics such as penicillins against Streptococci, Enterococci and some other organisms and this combination is used as first line therapy in enterococcal endocarditis.14 Aminoglycosides are inactive against anaerobes and Burkholderia cepacia.
Although these drugs are highly effective, their utilization has been limited due to concerns about (reversible) nephrotoxicity, ototoxicity and subsequent need for frequent monitoring
Mechanism of Action
Aminoglycosides are rapidly bactericidal and are known to bind irreversibly to the 30 S ribosomal subunit and alter protein synthesis.15
Clinical Indications and Dosage
Aminoglycosides are generally administered in combination with agents active against gram-positive and anaerobes for treatment of severe sepsis caused by enterobacteria and some other gram-negative aerobic bacilli. Others include endocarditis, respiratory and urinary tract infections and tuberculosis. Though there is no clear choice on grounds of toxicity because of no proven clinical relevance between the various members of the groups, gentamicin is a sensible choice in patients without renal functional deficit unless resistance is a major problem.
Tobramycin may have some advantage for proven P. aeruginosa or Acinetobacter infections. Amikacin is preferred if there is resistance to other aminoglycosides, and netlimicin is preferred in patients with auditory or vestibular deficits or established or impending renal insufficiency, including the elderly.14
Empirical maintenance doses for gentamicin and tobramycin range from 1.2 to 1.5 mg/kg every 8 hours and for amikacin 7.5 mg/kg every 8 to 12 hours. Clinical scenarios when dosage reductions may be necessary include advanced age, decreased renal blood flow, intrinsic renal disease and azotemia. Higher drug dosages may be required in neonates, burn patients, with serious pseudomonal infections, or in patients with cystic fibrosis.
A new strategy for dosing aminoglycosides is being used by many clinicians. A single large daily dose of aminoglycosides or 'once daily' aminoglycosides may be more effective than (or at least as effective as) and less toxic than the traditional divided daily doses. However, in patients with high creatinine clearance estimates (i.e., > 100 mL/min) serum monitoring is recommended when the dosing interval may need to be shortened to every 12 hours.15
Polymyxins
The polymyxins also known as cationic polypeptides consist of 5 chemically different compounds (polymyxins A-E). Polymyxin B and polymyxin E (colistin) are two clinically used members of this class.16 Originally introduced into clinical practice about 60 years ago as an agent active against gram-negative bacteria, polymyxins remained largely unused because of nephrotoxicity, neurotoxicity and ototoxicity associated with its use.17-19
However, the emergence of gram-negative bacteria resistant to most classes of commercially available antibiotics, including carbapenem and the shortage of new antimicrobial agents with activity against these bacteria has led to the reconsideration of polymyxins as a valuable therapeutic option.
Their activity spectrum includes only gram-negative aerobic bacilli, particularly non-fermentative gram-negative bacilli (P. aeruginosa and A. baumannii), and strains resistant to almost all commercially available antibiotics are still susceptible to polymyxins.20
Pseudomonas mallei, Burkholderia cepacia, Proteus spp., Providencia spp., Serratia spp., Edwardsiella spp. and Brucella spp. are all resistant to colistin.
In addition, colistin is not active against gram-negative and gram-positive aerobic cocci, gram-positive aerobic bacilli, all anaerobes, fungi, and parasites.21
Mechanism of Action
The polymyxins have a narrow spectrum of activity, mainly against non-fermentative gram-negative bacteria. Their bactericidal activity involves an initial electrostatic interaction between these cationic polypeptides and the negatively charge lipopolysaccharide (LPS) in the outer membrane of these bacteria, leading to displacement of calcium (Ca2+) and magnesium (Mg2+) which stabilize the LPS membrane, thus causing derangement of the cell membrane. This leads to disruption of the outer membrane, allowing the antibiotic to gain access to the cytoplasmic membrane beneath; resulting in cell lysis and death16,21
In addition to the direct antibacterial activity, colistin also has potent anti-endotoxin activity. The endotoxin of gram-negative bacteria is the lipid A portion of LPS molecules, and colistin binds and neutralizes LPS. The significance of this mechanism for in vivo antimicrobial action, namely prevention of the endotoxin's ability to induce shock through the release of cytokines, is not clear, because plasma endotoxin is immediately bound by LPS-binding protein, and the complex is quickly bound to cell surface CD14.21
Clinical Indications and Dosage
Intravenous polymyxins should be considered for the treatment of infections caused by Gram-negative bacteria resistant to other available antimicrobial agents, confi rmed by appropriate in vitro susceptibility testing.
The dosage of intravenous colistin recommended by the manufacturers in the USA Is 2.5-5 mg/kg (31,250-62,500 IU/kg) per day, divided into two to four equal doses (1 mg of colistin equals 12,500 IU). This dosage refers to adult patients with normal renal function.22
The dosage recommended by the manufacturers in the UK is 4-6 mg/kg (50,000- 75,000 IU/kg) per day, in three divided doses for adults and children with body weights of <60 kg and 80-160 mg (1-2 million IU [MIU]) every 8 hours for those with body weights of >60 kg.23 However, recent data does suggest the use of doses up to 3 MIU every 8 hours.24
Modifications of the total daily dose are required in the presence of renal impairment. For serum creatinine levels 1.3-1.5 mg/dL, 1.6-2.5 mg/dL or ≥2.6 mg/dL, the recommended dosage of intravenous colistin is 2 MIU every 8 hours, 12 hours or 24 hours, respectively.
In patients undergoing renal replacement therapy, the recommended dose is 2 MIU after each haemodialysis session and 2 MIU daily during peritoneal dialysis.24
Glycopeptides
The glycopeptide antibiotics, vancomycin and teicoplanin, have remained as the drugs of last resort for more than 20 years.
The spectrum of glycopeptides covers essentially the gram-positive organisms and a few anaerobes, and their activity against gram-negative organisms is most often marginal. In vitro pharmacodynamic models show that vancomycin exhibits timedependent bactericidal activity against most gram-positive organisms. However, vancomycin is essentially bacteriostatic against enterococci but may become bactericidal when combined with an aminoglycoside.25
Teicoplanin is a glycopeptide antibiotic with activity similar to vancomycin, except against streptococci, which are more susceptible to teicoplanin. It is not commercially available in the United States.25 The most striking difference between vancomycin and teicoplanin is their capacity to bind serum proteins, which is much higher for teicoplanin than for vancomycin. This explains the prolonged half-life of teicoplanin. Teicoplanin, because of its lipophilic character, is characterized by a higher volume of distribution than vancomycin, allowing it to reach therapeutic concentration in fat, muscles including pericardium and myocardium and to some extent, bone and cartilage, but not in the CNS.25
Mechanism of Action
Glycopeptides exhibit bactericidal activity by inhibition of bacterial cell wall synthesis. Specifically, it complexes with the D-alanyl-D-alanine portion of peptide precursor units, inhibiting peptidoglycan polymerase and transpeptidation reactions. This prevents cross-linking of the cell wall peptidoglycan, which occurs during the second stage of cell wall synthesis. Because beta-lactams inhibit cell wall biosynthesis in the third phase, there is no cross-resistance between the drugs, and no competition for binding sites.25
Clinical Indications and Dosage
Glycopeptides are the drug of choice for methicillin-resistant strains of coagulase-negative and coagulase-positive staphylococcal infections, including bacteraemia, endocarditis, pneumonia, cellulitis, and osteomyelitis. When patients are allergic to semisynthetic penicillins or cephalosporins, vancomycin is an alternative for methicillin-susceptible staphylococcal infections. In the case of serious infections caused by methicillin-susceptible organisms, such as endocarditis, however, vancomycin may be less effective than semisynthetic anti-staphylococcal penicillins and should not be used for convenience alone.26
The recommended dose of vancomycin is 500 mg q6h or 1g q12h27 and teicoplanin is three doses of 400 mg administered 12h apart, followed by a loading dose of 400 mg daily.28
Linezolid
Linezolid is the first of a truly new class of antibiotics, the oxazolidinones. The antibacterial spectrum of linezolid includes gram-positive pathogens and some gram negative anaerobic species but not gram-negative aerobes. Importantly, multi-drug resistant organisms such as methicillin-resistant staphylococci, staphylococci with reduced susceptibility to vancomycin, penicillin- and macrolide-resistant pneumococci and vancomycin-resistant enterococci (VRE) are fully susceptible to linezolid. Linezolid has almost 100% bioavailability and the area under the plasma concentration curve is identical after oral and i.v. administration. This enables initial oral administration of linezolid in those patients who can absorb the drug normally and also an early stepdown therapy from i.v. to oral.29
Mechanism of Action
Although, linezolid blocks protein synthesis as many other antibiotics such as macrolides, aminoglycosides, tetracyclines, streptogramins, chloramphenicol and lincosamides, the mode of action of linezolid is unique as the drug inhibits protein synthesis at a very early phase. Binding of linezolid to the 50S subunit of the ribosome prevents assembly of the ribosome and thereby formation of a functional initiation complex consisting of ribosome, tRNA and mRNA. Cross resistance to currently used antibiotics has not been observed.30
Clinical Indications and Dosage
Linezolid is approved for complicated SSSIs (cSSSIs), CAP including concurrent bacteraemia, nosocomial pneumonia, VREF including bacteraemia and uncomplicated skin and skin structure infections. The recommended adult dose is 400 or 600 mg i.v./oral twice a day.31
Despite the advantage of linezolid in the treatment of infections caused by MRSA in some studies, there are also reports of safety concerns. In particular, myelosuppression, especially thrombocytopenia, has been observed in 1-13% of patients after longer admission. In addition, linezolid treatment can cause peripheral and optic neuropathy with prolonged use (>28 days). Serotonin toxicity was reported in patients who received concomitant treatment with a selective serotonin-reuptake inhibitor.30
Lipopeptides
Daptomycin is the first of a new class of antibacterials, the cyclic lipopeptides, derived from Streptomyces roseosporus. In vitro studies demonstrated that daptomycin had bactericidal activity equal to or greater than that of vancomycin, linezolid and quinupristin-dalfopristin. Daptomycin is bactericidal against MRSA, MRSE and VRE, inclusive of linezolid-resistant isolates. However, recent studies reported daptomycinnonsusceptible VISA strains in patients with bacteraemia and endocarditis. Obviously, there is a correlation between vancomycin MICs and daptomycin susceptibility.30
Mechanism of Action
Daptomycin has a unique mechanism of action. The drug is postulated to undergo calcium-dependent oligomerisation and binding to the bacterial cell membrane, without penetration into the cytoplasm. The resulting alteration in the bacterial cell membrane leads to efflux of potassium from the cell and subsequent membrane depolarisation. This is thought to result in impairment of potassium-dependent macromolecular synthesis leading to cell death. In comparison to other antibacterial agents, daptomycin in vitro has shown to result in minimal release of bacterial toxins into the circulation. Although the exact mechanism is unknown, it is possible that fewer pro-infl ammatory bacterial fragments are released by the bactericidal action of daptomycin.32
Clinical Indications and Dosage
Daptomycin had been approved in 2003 for use in soft-tissue infections, bacteraemia and right-sided endocarditis. It is not recommended for treatment of pneumonia owing to its inhibition by pulmonary surfactant.30
The recommended dose of daptomycin is 4 mg/kg and 6 mg/kg in cSSSIs and S. aureus bloodstream infections (bacteraemia), including those with right-sided endocarditis, caused by methicillin-susceptible and methicillin-resistant strains, respectively.
Because daptomycin is eliminated primarily by the kidney, a dosage modifi cation is recommended for patients with creatinine clearance <30 mL/min, including patients receiving haemodialysis or CAPD. In Phase 1 and 2 clinical studies, CPK elevations appeared to be more frequent when daptomycin was dosed more frequently than once daily. Therefore, daptomycin should not be dosed more frequently than once a day.33
Glycylcycline
Tigecycline is the first glycylcycline and the first new tetracycline analogue since minocycline over 30 years ago. It is also unique among the small raft of anti-MRSA and anti-enterococcal drugs now reaching the market because it additionally has substantial anti-gram-negative activity, encompassing not only most Enterobacteriaceae, but also 'at least in vitro' multiresistant A. baumannii.34
Although tigecycline is structurally similar to tetracyclines, it is not a substrate for Tet A-E efflux pumps, and, therefore, also effective against most Enterobacteriaceae and A. baumannii that acquired tetracycline resistance. However, tigecycline does not exhibit potent antibacterial activity against P. aeruginosa since it is substrate for MexXY efflux pumps.30
Mechanism of Action
Tigecycline inhibits protein synthesis in a broad range of bacteria by binding to the 30S ribosomal subunit with fi ve times higher affinity than tetracycline, blocking entry of amino-acyl tRNA molecules into the A site of the ribosome. This prevents incorporation of amino acid residues into elongating peptide chains, inhibiting protein synthesis and bacterial growth across a broad spectrum of pathogens.30
Clinical Indications and Dosage
It is approved for the treatment of skin and soft tissue infections, intra-abdominal infections, CAP and is currently being evaluated for the treatment of nosocomial pneumonia.
The recommended dose is 100 mg as an initial loading dose, followed by 50 mg every 12 hours. For patients with severe hepatic impairment (Child-Pugh C), the recommended dose is 100 mg as an initial loading dose, followed by 25 mg every 12 hours. No dosage adjustments are needed in patients with mild-to-moderate hepatic or renal impairment. Tigecycline is not removed by haemodialysis. The use of tigecycline in patients less than 18 years of age is not recommended since no usage data are available.35
Quinupristin/Dalfopristin
Quinupristin/dalfopristin (Q/D) i.v., a streptogramin antibacterial agent, is a formulation of two semisynthetic pristinamycin derivatives, quinupristin and dalfopristin in the ratio of 30:70 (w/w). It belongs to the macrolide-lincosamide-streptogramin group of antibiotics that exerts potent in vitro activity against gram-positive organisms most frequently encountered in cSSSIs, including S. aureus and S. pyogenes. In addition, Q/D has activity against most methicillin-, lincosamide- and erythromycin-resistant strains of coagulase-negative staphylococci and S. aureus. The drug is also active against glycopeptide-resistant (vancomycin- and teicoplanin-resistant) S. aureus (VISA) and vancomycin-resistant Enterococcus faecium but not against vancomycin-resistant E. faecalis.
Presently, Q/D has not gained approval by the FDA for the treatment of MRSA infections.30
Mechanism of Action
The combination of Q/D is synergistic, and is generally bactericidal compared with either agent used alone or compared with similar antibiotics in the macrolide group. The main target is the bacterial 50S ribosome, with the formulation acting to inhibit protein synthesis. Dalfopristin blocks an early step in protein synthesis by forming a bond with a ribosome to prevent elongation of the peptide chain. Quinupristin blocks a later step by preventing the extension of peptide chains and causing incomplete chains to be released.36
Cross-resistance has not been reported between Q/D and glycopeptide, quinolone or β-lactam antimicrobials.30
Clinical Indications and Dosage
Q/D was approved by the US FDA in 1999 to be the first antibacterial drug to treat infections associated with vancomycin-resistant E. faecium bacteraemia (VREF) when no alternative treatment is available and also for cSSSIs.
Q/D should be administered by intravenous infusion in 5% Dextrose in Water solution over a 60-minute period. The recommended dosage of Q/D for the treatment of VREF and cSSSIs in adults is 7.5 mg per kg administered intravenously every eight hours and 7.5 mg per kg given intravenously every 12 hours respectively. The dosage of Q/D does not have to be adjusted in elderly patients or patients with renal impairment. Limited clinical data are available on the use of Q/D in patients with hepatic disease, but a dosage reduction may be required in patients with cirrhosis. The safety and effi cacy of Q/D have not been well studied in children.37
In vitro drug interaction studies have demonstrated that Q/D significantly inhibits CYP450 3A4 metabolism of cyclosporin A, midazolam, nifedipine and terfenadine. Therefore, co-administration of Q/D with drugs that are CYP450 3A4 substrates and possess a narrow therapeutic window requires caution and monitoring of these drugs (e.g., cyclosporine).30
Ceftobiprole
Ceftobiprole is a novel broad-spectrum cephalosporin with activity against a wide range of gram-positive and gram-negative bacteria, including several resistant species such as methicillin-resistant S. aureus and penicillin-resistant Streptococcus pneumoniae. Ceftobiprole is administered intravenously as the prodrug ceftobiprole medocaril, which is converted by plasma esterases to ceftobiprole in <30 minutes.38,39 It is approved in Canada under the trade name ZefteraTM.
Like cefotaxime, ceftriaxone, ceftazidime and cefepime, ceftobiprole possesses limited activity against anaerobes such as Bacteroides fragilis and non-fragilis Bacteroides spp. In vitro resistance-development studies indicated that ceftobiprole has a low propensity to select for resistant subpopulations. Ceftobiprole has a modest postantibiotic effect (PAE) of approximately 0.5 h for MRSA.30
Clinical Indications and Dosage
It is currently approved in Canada (Zevtera) for cSSSIs, including non-limb-threatening diabetic foot infections without osteomyelitis.
The antibiotic is currently undergoing Phase 3 clinical trials for hospital-acquired pneumonia, CAP requiring hospitalization, and fever and neutropenia in patients receiving chemotherapy.
Based on pharmacokinetic and pharmacodynamic data, Monte Carlo simulations, and Phase 3 clinical trials, the predicted FDA-approved dose of ceftobiprole medocaril for suspected or documented gram-postive cSSSIs will be 500mg intravenously every 12 hours (1-h infusion) for 7-14 days. Predicted approved dosing if gram-negative pathogens are suspected or isolated, such as in a diabetic foot infection, may be 500 mg intravenously every 8 hours (2-h infusion) for 7-14 days.40
Quinolones
The evolution of quinolones actually emanated from the discovery of nalidixic acid in 1962 as a by-product of antimalarial research, the first representative of the quinolones which was found effective against some gram-negative bacteria.41 Nalidixic acid had several weaknesses, but it did provide the basis for modifi cation into more favorable analogues. Placement of a fl uorine atom at the 6 position of the quinolone nucleus and replacement of the 7-methyl side-chain of nalidixic acid with a piperazine group led to development of ciprofl oxacin and ofl oxacin with excellent gram-negative activity including P. aeruginosa, but weak gram-positive activity.42
The new respiratory fluoroquinolones (gatifl oxacin, gemifl oxacin, levofl oxacin, moxifl oxacin, and on the horizon, garenoxacin) off er many improved qualities over older agents such as ciprofl oxacin. These include retaining excellent activity against gram-negative bacilli, with improved gram-positive activity (including Streptococcus pneumoniae and S. aureus). In addition, gatifl oxacin, moxifl oxacin and garenoxacin all demonstrate increased anaerobic activity (including activity against Bacteroides fragilis). The new fluoroquinolones possess greater bioavailability and longer serum half-lives compared with ciprofl oxacin. The new fluoroquinolones allow for once-daily administration, which may improve patient adherence. The high bioavailability allows for rapid step down from intravenous administration to oral therapy, minimizing unnecessary hospitalization, which may decrease costs and improve quality of life of patients.43
Pazufloxacin mesylate is a novel 3rd generation injectable quinolone, having 1-amino cyclopropyl group inserted via C - C (Carbon-Carbon bond) to the 7 position that is not seen in the existing new quinolone antibacterial drugs. An oxazine ring is said to provide a potent broad spectrum activity even against resistant bacteria e.g. MRSA; ampicillin-resistant H. influenzae; ESBLs producing E. coli and Klebsiella; and imipenemresistant P. aeruginosa.44
Mechanism of Action
Quinolones rapidly inhibit DNA synthesis by promoting cleavage of bacterial DNA in the DNA-enzyme complexes of DNA gyrase and type IV topoisomerase, resulting in rapid bacterial death.
Clinical Indications
Quinolones are clinically useful in varied indications45 as listed below:
- Respiratory tract infections (i.e. acute bacterial exacerbations of chronic bronchitis, CAP and sinusitis)
- Uncomplicated and some complicated urinary tract infections
- Bacterial prostatitis
- Skin and soft tissue infections, bone and joint infections, gastrointestinal infections, particularly infectious diarrhoea caused by toxigenic E. coli, Salmonella enteric, Shigella, Aeromonas and Vibrio species
- Sexullay transmitted diseases (gonococcal and chamydial infections; chancroid) and pelvic infections
Macrolides
Erythromycin, the first macrolide antibiotic to be available in clinical practice, is characterized by poor water solubility and rapid inactivation by stomach acidity, which results in widely varying bioavailability after oral administration. Structural alterations of erythromycin A have resulted in improved pharmacokinetic properties, including bioavailability, gastrointestinal tolerance, higher peak plasma levels, longer apparent elimination plasma half-lives and improved tissue concentrations.
The most important therapeutic macrolides are characterized by 14-, 15- or 16- membered lactone ring and share the same antibacterial spectrum, including most gram-positive organisms, Neisseria spp., Haemophilus spp., Bordetella pertussis, Moraxella catarrhalis and both gram-positive and gram-negative anaerobes.
The semisynthetic macrolides (clarithromycin, azithromycin) exert important activity against intracellular pathogens, including Chlamydia trachomatis, Chlamydia pneumonia, Legionella pneumophila and other Legionella spp., the Mycobacterium avium complex. Azithromycin appears to be most active against atypical pathogens.
They are inactive or poorly active against Enterobacteriaceae and non-fermentative gram-negative bacteria such as P. aeruginosa.46
Trimethoprim-Sulfamethoxazole
The introduction of this combination constitutes an important advance in the development of clinically effective antimicrobial agents and represents the practical application of a theoretical consideration i.e., if two drugs act on sequential steps in the pathway of an obligate enzymatic reaction in bacteria, the result will be synergistic. In much of the world, the combination is known as cotrimoxazole. The spectrum of trimethoprim is similar to that of sulfamethoxazole, although the former is 20 to 100 times more potent than latter. Most gram-positive and gram-negative microorganisms are sensitive to trimethoprim; however resistance can develop when the drug is used alone.
From 50-95% of strains of S. aureus, S. epidermidis, S. pyogenes, the viridians group of streptococci, E. coli, Proteus mirabilis, Proteus morganii, Proteus rettgeri, Enterobacter spp., Salmonella, Shigella, Pseduomonas pseudomallei, Serratia and Alcaligenes spp. are inhibited. Methicillin-resistant S. aureus although resistant to trimethorpim or sulfamethoxazole alone, may be susceptible to combination. The combination appears to have special effi cacy in chronic and recurrent infections of urinary tract and bacterial rostatitis.47
Chloramphenicol
With its introduction in 1948, it became evident that chloramphenicol could cause serious and fatal blood dyscrasias. For this reason, its use was reserved for treatment of serious life threatening infections in patients who cannot take safer alternatives because of resistance or allergies. Chloramphenicol inhibits protein synthesis in bacteria, and to a lesser extent, in eukaryotic cells.48
Chloramphenicol possesses a broad spectrum of antimicrobial activity to include S. aureus, Streptococccus pyogens, Streptococcus pneumoniae, Enterococcus faecalis, Neisseria gonorrhoeae. Most gram negative bacilli are susceptible, but P. aeruginosa is resistant. It is strictly bacteriostatic against almost all bacterial species, but exerts a bactericidal eff ect 2-4 times the minimum inhibitory concentration against some strains of gram-positive cocci, H. influenzae and Neisseria spp. The minimum bactericidal concentrations of chloramphenicol for penicillin-resistant pneumococci are often significantly higher than those for penicillin-susceptible strains. Its bacteriostatic effect may inhibit the action of penicillins and other beta-lactam antibiotics against Klebsiella penumoniae and other enterobacteria in vitro, but the clinical significance of this is doubtful.49
Tetracyclines
Chemically, the tetracyclines are naphthacene derivatives, made up by fusion of four partially unsaturated cyclohexane radicals and hence the name tetracyclines. Various tetracyclines have similar antibacterial spectrum and differ only in structure. They are bacteriostatic and along with chloramphenicol are termed broad spectrum antibiotics, as in addition to their antibacterial activity against a number of gram-positive and gram-negative organisms, they also inhibit the growth of certain actinomyces (fungi), rickettsiae and chlamydia organisms. Gram-positive organisms in general respond better than gram-negative organisms.50
Fosfomycin
Fosfomycin is a broad-spectrum antibacterial with activity against gram-positive and gram-negative bacteria. It has in vitro activity against S. aureus, S. epidermidis, Streptococcus pneumoniae, and E. faecalis. Listeria monocytogenes is resistant to fosfomycin, whereas other Listeria species (e.g., Listeria ivanovii) may be susceptible.
Fosfomycin shows very good activity against many gram-negative bacteria, such as E. coli, Proteus spp., Klebsiella pneumoniae, Enterobacter spp., Serratia marcescens, and Salmonella typhi.
P. aeruginosa and Bacteroides fragilis exhibits considerable rates of resistance. However, a combination of fosfomycin and beta-lactam (cefepime, aztreonam, or meropenem) was effective in an in vitro study involving P. aeruginosa clinical isolates from sputum, urine, and blood samples. A. baumannii is resistant to fosfomycin, although in vitro combinations of fosfomycin with aminoglycosides may be synergistic.51
The oral form of this broad-spectrum antibiotic has principally been used in the treatment of uncomplicated urinary tract infections (UTIs) in the USA, the UK and other countries. However, the intravenous form has been used for indications beyond UTIs in only a few countries such as Germany, France, Spain and Japan.52
Nitrofurans
While several thousand nitrofuran compounds have been synthesised, only two derivatives have become established in clinical use: nitrofurazone and nitrofurantoin.
The nitrofurans have remained clinically useful against a wide spectrum of grampositive and gram-negative bacteria, including many strains of common urinary tract pathogens.
It appears that the nitrofurans act by inhibiting bacterial enzymes involved in DNA and RNA synthesis, carbohydrate metabolism, and other metabolic enzyme proteins.Organic matter (e.g. blood and serum) slightly reduces the antibacterial activity of nitrofurazone in vitro, but this small reduction is not likely to be clinically important in patients with burns or other surface wounds containing blood or serum. Aminobenzoic acid also has an inhibitory eff ect on the antibacterial activity of nitrofurazone.
Nitrofurazone is primarily used as a topical antibacterial agent in patients with burns and skin grafts, wound infections, but it has also been of clinical utility in vaginal surgery, external ocular infections, insect bites, stasis ulcers, bacterial ear infections, and middle ear surgery and recently was approved for the prophylaxis of catheterassociated urinary tract infection (CAUTI).53
Nitrofurantoin, introduced initially as microcrystalline form, is now also available as macrocrystalline form with improved gastrointestinal tolerance. It has a terminal disposition half-life of approximately 20 minutes with 40% of the drug being excreted into the urine in the therapeutically active unchanged form.53
It is currently available as capsules, tablets and oral suspension and is prescribed for two indications: as defi nitive therapy for established UTIs and as a prophylactic agent for patients with recurrent UTIs.54
Lincosamides
Lincosamides (clindamycin and lincomycin) have an unusual antimicrobial spectrum, being widely active against gram-positive bacteria (including methicillin-resistant strains) and most anaerobes. They are also active against some mycoplasmas and protozoa, but not against gram-negative aerobes and enterococci. They are clinically useful in penicillin-susceptible infections in allergic patients, staphylococcal infections, particularly bone and joints, and anaerobic infections. They are widely distributed in various tissues, including penetration into cells and bone. They are generally well tolerated, except for the relative frequency with which they have been associated with severe diarrhoea, including Clostridium difficle-associated pseudomembranous colitis.55
5-Nitroimidazoles
Among 5-nitroimidazoles, metronidazole, tinidazole and ornidazole are in widespread clinical use. Others include secnidazole and nimorazole, while carnidazole, dimetridazole, ipronidazole and ronidazole are used in veterinary medicine. They exhibit excellent potency against common anaerobic pathogens as well as several microaerophilic species, including Helicobacter pylori and Gardnerella vaginalis. Susceptible protozoa include Giardia lamblia, Entamoeba histolytica, T.vaginalis, Balantidium coli and Blastocystis hominis.56
Anti-Fungals
Antifungal agents are classifi ed, based on their proposed mechanism of action and chemical structure, as follows:
- Plasma membrane function/synthesis inhibitors - it includes agents like Allylamines (e.g. terbinafi ne), Polyenes (e.g. amphotericin B) and Azoles (e.g. voriconazole, itraconazole, posaconazole)
- Agents that inhibit the cell wall synthesis, including Echinocandins (caspofungin, anidulafungin, micafungin)
- Agents that inhibit nucleic acid synthesis, including flucytosine
- Agents that inhibit cell division (e.g. griseofulvin)
Flucytosine
Flucytosine (5-flucytosine, 5-FC, Ancobon) is an antifungal agent originally developed in 1957 as an antimetabolite. Although it has found no role as an anti-tumor agent, it is used for the treatment of certain fungal infections.
Due to a high incidence of primary and/or acquired resistance, use of flucytosine as monotherapy is significantly restricted. Combination therapy of 5-FC with amphotericin B is recommended for the initial management of severe cryptococcal pneumonia and meningoencephalitis infections and (less frequently) in select invasive candidal infections.57
Mechanism of Action
5-FC exerts its antifungal effects by interfering with both DNA and protein synthesis. 5-FC is transported into susceptible fungi by cytosine permease, then deaminated to 5-fluorouracil (5-FU) by cytosine deaminase. The absence of cytosine deaminase in mammalian cells allows selective effects on fungal cells.57
Polyenes
Amphotericin B (AMB) and nystatin are the currently available polyenes, although differing safety profi les have limited nystatin to topical use. The broad antifungal spectrum and experience with the use of amphotericin B accounts for its continued use despite toxicity concerns.
AMB is primarily used intravenously or through the inhalational route. In attempts to avoid the nephrotoxicity seen with amphotericin B deoxycholate, several other formulations have been developed. The lipid preparations include: liposomal amphotericin B (L-AMB), amphotericin B lipid complex, (ABLC) and amphotericin B colloidal dispersion (ABCD).
All currently available formulations are highly protein bound (>95%, primarily to albumin) and have long half-lives.58
Mechanism of Action
The polyenes bind to ergosterol present within the fungal cell wall membrane. This process disrupts cell wall permeability by forming oligodendromes functioning as pores with the subsequent efflux of potassium and intracellular molecules causing fungal death. There is also evidence that AMB acts as a proinfl ammatory agent and further serves to stimulate innate host immunity.58
Clinical Indications and Dosage
AMB exhibits poor cerebrospinal fluid levels (<5% of concurrent serum concentration); however, this agent remains the treatment of choice for cryptococcal meningitis. The recommended dose for conventional amphotericin B is 0.3-1.5 mg/kg infused over 1-4 hours, and for liposomal amphotericin B is 3-5 mg/kg once daily, except for cryptococcal meningitis in HIV where the dose is 6 mg/kg once daily infused over 120 minutes.58
Azoles
The azoles that are available for systemic use can be classifi ed into two groups: the triazoles (fl uconazole, itraconazole, voriconazole, posaconazole) and the imidazoles (ketoconazole).
Agents within the azole class vary importantly with regards to spectrum of activity, pharmacokinetic profi les, and toxicities. For example, fl uconazole has excellent activity against yeasts, but off ers no protection against mold infections. An extended spectrum is provided by itraconazole, but inconsistent bioavailability limits use of this agent in severely ill patients. Voriconazole is the first-line agent for the treatment of invasive aspergillosis, but it is associated with unique side effects, and lacks activity against the Mucorales, the agents of mucormycosis. Posaconazole has the broadest spectrum of activity and fewest drug interactions, but absorption is problematic and it is available only as an oral formulation.59
Mechanism of Action
Azoles exhibit their antifungal eff ect by inhibiting the fungal cytochrome enzyme, 14-alpha-sterol demethylase involved in ergosterol biosynthesis, thus inhibiting fungal cell growth.59
Echinocandins
The echinocandins are large lipopeptide molecules that are inhibitors of β-(1, 3)-glucan synthesis, an action that damages fungal cell walls. In vitro and in vivo, the echinocandins are rapidly fungicidal against most Candida spp. and fungistatic against Aspergillus spp. They are not active at clinically relevant concentrations against Zygomycetes, Cryptococcus neoformans, or Fusarium spp. No drug target is present in mammalian cells. The first of the class to be licensed was caspofungin, for refractory invasive aspergillosis (about 40% response rate) and the second was micafungin, followed by anidulafungin. The echinocandins are widely distributed in the body, and are metabolized by the liver.60
Activity is comparable between the three agents, although limited data indicate that anidulafungin may have low MICs against C. parapsilosis and Candida glabrata strains that demonstrate elevated MICs to caspofungin and micafungin. All three drugs have good fungistatic activity against Aspergillus spp., although minimal effective concentrations of micafungin and anidulafungin are 2- to 10-fold lower than those for caspofungin. Synergistic/additive in vitro effects of echinocandins when combined with a polyene or azole have been observed.
All echinocandins have low oral bioavailability, and distribute well into tissues, but poorly into the CNS and eye. Anidulafungin is unique in that it undergoes elimination by chemical degradation in bile rather than via hepatic metabolism, has a lower maximum concentration and smaller steady state under the concentration-time curve but longer half-life than caspofungin or micafungin.
Clinical Indications and Dosage
All three agents are approved for the treatment of oesophageal candidiasis, candidaemia and other select forms of invasive candidiasis. Only micafungin is licensed for antifungal prophylaxis in stem cell transplantation, whereas caspofungin is approved for empirical therapy of febrile neutropenia. Caspofungin has been evaluated in the salvage and primary therapy of invasive aspergillosis. The recommended dose of caspofungin is 70 mg once daily as a loading dose, followed by 50 mg once daily, while for anidulafungin loading dose of 200 mg on day one, followed by 100 mg daily is recommended. No loading dose is required for micafungin and the recommended dose is 50-150 mg once a day.61
Terbinafine
Terbinafine is an allylamine that has a broad spectrum of activity against fungal pathogens of the skin, hair and nails, including dermatophytes such as Trichophyton (e.g., T. rubrum, T. mentagrophytes, T. verrucosum, T. tonsurans, and T. violaceum), Microsporum (e.g., M. canis), Epidermophyton fl occosum, and yeasts of the genera Candida (e.g., C. albicans) and Pityrosporum.
Mechanism of Action
Terbinafi ne interferes specifi cally with fungal sterol biosynthesis at an early stage. This leads to a defi ciency in ergosterol and to an intracellular accumulation of squalene, resulting in fungal cell death. Terbinafi ne acts by inhibition of squalene epoxidase in the fungal cell membrane. The enzyme, squalene epoxidase, is not linked to the cytochrome P-450 system. Terbinafi ne does not infl uence the metabolism of hormones or other drugs.
Clinical Indications and Dosage
When given orally, the drug concentrates in skin, hair and nails at levels associated with fungicidal activity. One tablet of 250 mg once a day for fungal infections of the skin, including the treatment of tinea corporis, tinea cruris, tinea pedis, and yeast infections of the skin caused by the genus Candida (e.g., Candida albicans) where oral therapy is generally considered appropriate, owing to the site, severity or extent of the infection
Aerosolized Antibiotics
Aerosol administration of antibiotics has gained increasing attention for the treatment of pulmonary infections, particularly chronic pulmonary infections in cystic fibrosis (CF) patients. This route of administration can produce local concentrations of drug far in excess of those that can be obtained with conventional dosing. These concentrations can improve the antibacterial killing of existing agents against organisms considered resistant by regulatory or consensus susceptibility breakpoints, and achieve pharmacokinetic pharmacodynamic (PK PD) targets against these same organisms that are associated with bacterial killing and reduced potential for resistance.62
Tobramycin was the first antibiotic which was US FDA in the year 1997, followed by aztreonam in 2010 for pulmonary infections in cystic fibrosis (CF) patients. Similar to the aminoglycosides, inhalation is a potentially attractive route of colistin administration given the toxicities observed with systemic administration. Inhaled amphotericin B was the first drug used for the treatment of systemic mycosis, and earlier animal experiments using aerosolized amphotericin B showed favorable effi cacy in the treatment of pulmonary aspergillosis. Others under development are amikacin, levofl oxacin, ciprofl oxacin and vancomcyin.63
Anti-Viral Agents
The antivirals are classified as follows:64
- Anti-herpes virus agents (e.g. acyclovir, ganciclovir, valacyclovir)
- Anti-infl uenza virus agents (e.g. amantadine, oseltamivir, ribavirin)
- Anti-hepatitis virus B agents (e.g. lamivudine, adefovir)
- Anti-HIV agents (e.g. zidovudine, nevirapine, efavirenz, lopinavir)
Acyclovir
Acyclovir is a prototype of antiviral agents. Herpes simplex virus (HSV)-types 1 and 2, Varicella-Zoster virus (VZV), and some Epstein-Barr virus-mediated infections are sensitive to acyclovir. It can be administered by oral, topical or intravenous routes.65
Mechanism of Action
Acyclovir is phosphorylated intracellularly by a viral kinase and subsequently by the host cell. This triphosphate analog causes viral inhibition by interfering with DNA polymerase, leading to inhibition of DNA synthesis.65
Clinical indications and dosage
Acyclovir is recommended for the treatment of:
- Herpes simplex infections in immunocompromised patients
- Herpes simplex encephalitis
- Neonatal Herpes simplex infection
- Varicella-zoster infections in immunocompromised patients
The usual dose of acyclovir in patients with Herpes simplex (except herpes encephalitis) or Varicella-zoster infections should be given acyclovir for infusion in doses of 5 mg/kg bodyweight every eight hours.
Immunocompromised patients with Varicella-zoster infections or patients with herpes encephalitis should be given acyclovir for infusion in doses of 10 mg/kg bodyweight every eight hours provided renal function is not impaired.
Dose adjustment is required in patients with renal impairment.66
Anti-influenza Virus Agents
Neuraminidase inhibitors (e.g. zanamivir, oseltamivir) are the agents that inhibit the enzyme neuraminidase, inhibiting the release of new virions and thus the spread of virus. These are effective against both, types A and B, of infl uenza virus.65
Amantadine and rimantadine are agents that inhibit the replication of infl uenza virus by inhibiting viral uncoating. They are recommended for the treatment of infections caused by Infl uenza A virus.67
Pharmacokinetic-Pharmacodynamic (PK-PD) considerations
Extensive research over the last decade has determined the PK PD relationships associated with antimicrobial effects against gram-positive and gram-negative bacteria. For most drugs, including aminoglycosides and fluoroquinolones, the AUC: MIC (area under the curve: minimum inhibitory concentration) ratio is associated with the extent of bacterial killing. In settings where emergence of resistance can occur (e.g. P. aeruginosa infections), high Cmax: MIC ratios can suppress the selection of resistant organisms for bactericidal agents.
In contrast, beta-lactam and monobactam agents have time-dependent antimicrobial effects; thus, the percent of a dosing interval that concentrations exceed the MIC (% T > MIC) is linked with antimicrobial effects.68 In vitro and in vivo animal studies suggest that penicillins require a %T>MIC of 50% for bactericidal activity and cephalosporins require a %T>MIC of 65% to 75%. In contrast, carbapenems require %T>MIC of 20% of the dosage interval to achieve a bacteriostatic eff ect and a %T>MIC of 40% for bactericidal activity.
As such increasing the dose while keeping the dosage interval the same, reducing the dosage interval (i.e., administering the drug more frequently) while keeping the dose fixed, or increasing the duration of i.v. infusion for parenteral agents would be required to achieve optimal antimicrobial effects.69
General Principles of Anti-Infective Therapy
A number of patient specifi c historical and clinical factors must be taken into account before an empirical antibiotic regimen is selected. For example, several aspects of the patient's history must be considered
- Is the infection community-acquired or hospital acquired?
Community-acquired infections are, in general, caused by bacteria that are more susceptible to a wide range of antimicrobial agents and may be treated with 'standard , less toxic and less expensive agents. Hospital-acquired (nosocomial) infections, however, may be caused by pathogens with complex antimicrobial resistance patterns and may require potentially more toxic and expensive drugs.
- What is the patient's underlying illness?
Certain diseases predispose patients to infections caused by specifi c pathogens. For example, multiple myeloma is associated with infections caused by encapsulated bacteria, such as S. pneumonia and H. influenzae, patients with leukaemia and leucopenia and burn victims are predisposed to infections with P. aeruginosa; and diabetes mellitus is associated with polymicrobial infections caused by S. aureus and anaerobic organisms. Thus, if a patient with such an underlying illness is thought to have an infection, the empirical antibiotic regimen is tailored to cover these organisms.
What is the suspected primary focus of infection?
Different anatomic sites are characteristically infected by specific microorganisms. For example, the urinary tract and biliary tract are usually infected by enteric (aerobic) gram-negative bacilli, whereas endocarditis is caused by aerobic grampositive cocci, such as S. aureus and viridians group streptococci, and meningitis is caused by encapsulated organisms, such as S. pneumoniae, Neisseria meningitidis, and H. influenzae.
Has the patient been treated with antibiotics recently?
If the patient has acquired a new infection during or shortly after treatment with antibiotics, the new infection or superinfection is like to be caused by a more resistant pathogen.
In patients already receiving antibiotics, bacterial growth obtained may be suppressed in cultures owing to the presence of antibiotic in the actual culture specimen. Thus, culture results may be less helpful for pathogen identification.33
The appropriate selection of antimicrobial agents for empirical therapy also requires a reasonable understanding of drug pharmacology, including the mechanism of action (bactericidal versus bacteriostatic), antimicrobial spectrum of activity, pharmacokinetics, potential toxicities, and appropriate dose and dosing interval for a given clinical scenario. There are several important considerations:
- When should a bactericidal versus a bacteriostatic antibiotic be selected? Bactericidal antibiotics kill bacteria without the assistance of host defenses and thus are important for the cure of infections when the host immune response is not adequate. Three clinical settings require bactericidal therapy for cure: (a) meningitis, because there are decreased antibodies and complement (C3b) in cerebrospinal fluid (CSF) as well as the lack of surface phagocytosis: (b) infectious endocarditis, in which a dease (fi brin-platelet meshwork encases large bacterial inoculums (vegetation) and is impermeable to the effects of circulating white blood cells (WBCs) and (c) infections in neutropenic patients.
- Bacteriostatic drugs, in contrast, inhibit the normal growth cycle of bacteria but do not lyse or kill the organisms. Bacteriostatic agents, such as tetracyclines, macrolides, and clindamycin generally provide adequate therapy when host defenses are intact.
- The general spectrum of activity for an antimicrobial agent must be understood. The usefulness of a specifi c agent in an institution may be monitored by the microbiology; infectious diseases, and infection control departments. An antibiogram, which outlines an institution's susceptibility pattern to specifi c microorganisms, is helpful for the empirical antimicrobial selection process. The ICU physician must be informed of susceptibilities of common ICU pathogens because they may vary among institutions and also within a given institution (e.g. Enterobacter spp., Acinetobacter spp., and Pseudomonas spp.)
- Pharmacokinetics is important because the physician must understand the distribution and elimination characteristics of an antimicrobial agent. For example, infections occurring in the CSF (i.e. meningitis), biliary tract, or urinary tract may be diffi cult to treat because certain drugs may not adequately distribute to those sites. The routes of elimination for each agent are important for the drug selection process to ensure adequate urine and bile concentrations for the treatment of infection at these sites. Also, toxicities may occur when elimination routes are impaired.
- The dose and dosing intervals are selected with the following factors in mind: the pathogen's MIC, the severity of infection, the body size of the patient (aff ecting the drug's volume of distribution) and the route of drug elimination relative to the patient's organ function. Inadequate drug regimens can result in treatment failure and the emergence of antimicrobial may result in toxicity and unnecessary expense.
- The toxicity profi le of antimicrobial agents is important for the overall clinical outcome. Factors that contribute to potential toxicities may be avoided or carefully monitored (i.e. excessive serum drug concentrations, renal dysfunction, and concomitant administration of drugs with similar toxicities).
During the first 48 to 72 hours of empirical antimicrobial therapy, the patient must be watched closely for signs of clinical deterioration or improvement. If the patient's status deteriorates, the following possibilities should be considered.
- Presence of an undrained or undebrided focus of infection
- A too-narrow spectrum of antimicrobial coverage
- Inadequate penetration of drug to the focus of infection
- Sub-therapeutic dose or interval for the severity of infection or for the patient's body size.
When culture and sensitivity results are available, the antimicrobial regimen should be reassessed and tailored to the narrowest spectrum, least toxic and least expensive agents. It is crucial to tailor antimicrobial agents, even when patients have 'responded' to therapy. This is done to minimize the alterations in the patient's bacterial fl ora, which occur with prolonged use of broad-spectrum antibiotics. Shortened courses of therapy and narrow-spectrum coverage to both help minimize the 'antibiotic pressure' that promotes antimicrobial resistance' and reduce the incidence of bacterial superinfections caused by resistant pathogens (e.g. fungi and Enterococcus spp.). Drug toxicities and expense may also be reduced by decreasing the duration of therapy.70
References
1. Henry F. Chambers. Chapter 42 General Principles of Antimicrobial Therapy. In Laurence L.
Brunton, John S.Lazo, Keith L. Parker, editors. Goodman and Gilman's: The Pharmacological Basics of
Therapeutics. McGraw-Hill Medical Publishing Division 2006; 1095-1110.
2. Pramod M. Shah1 and Robin D. Isaacs. Ertapenem, the first of a new group of carbapenems. J Antimicrob Chemother 2003; 52: 538-542.
3. Joan E. Kapusnik-Uner, Marjorie and Merle A. Sande. Chapter 58 Antimicrobial Therapy in the Critical Care Setting. In pp: 659-674.
4. R.S.Satoskar, S.D. Bhandarkar, Nirmala N. Rege, editors. Pharmacology and Pharmacotherapeutics, 20th ed 2007. Pp: 648-668.
5. K.Bush. Chapter 16 Beta-Lactam Antibiotics: penicillins. In Roger Finch, David Greenwood, S.Norrby and Richard Whitlety, editors. Antibiotic and Chemotherapy. Anti-infectives agents and their use in therapy 2003; pp: 224-258.
6. William A Petri. Chapter 44 Penicillins, Cephalosporins and Other Beta-lactam Antibiotics. In Laurence L. Brunton, John S.Lazo, Keith L. Parker, editors. Goodman and Gilman's. The Pharmacological Basics of Therapeutics. McGraw-Hill Medical Publishing Division 2006; 1127-1154.
7. Nelson Lee, Kwok-Yung Yuen and Cyrus R. Kumana. Clinical Role of ?-Lactam/?-Lactamase Inhibitor Combinations. Drugs 2003; 63(14): 1511-1524.
8. Pramod M. Shah1 and Robin D. Isaacs. Ertapenem, the first of a new group of carbapenems. J Antimicrob Chemother 2003; 52: 538-542.
9. Katherine M. Knapp and B. Keith English. Carbapenems. Semin in Pedia Infect Dis 2001; 12(3): 175-185.
10. George G. Zhanel, Ryan Wiebe,Leanne Dilay, Kristjan Thomson, Ethan Rubinstein, Daryl J. Hoban, Ayman M. Noreddin and James A. Karlowsky. Comparative Review of the Carbapenems. Drugs 2007; 67(7): 1027-1052.
11. Kristen N Sch-Ryan Wiebe, lames A KarIowsky Ethan Rubinstein, Dary J Hoban and George Gzhanelt. Faropenem: review of a new oral penem. Expert Rev Infect Ther 2007; 5(2): 185-198.
12. Sutep Jaruratanasirikul, Somchai Sriwiriyajan and Jarurat Punyo. Comparison of the Pharmacodynamics of Meropenem in Patients with Ventilator-Associated Pneumonia following Administration by 3-Hour Infusion or Bolus Injection. Antimicrob Agents Chemother 2005; 49 (4):1337-1339.
13. Zienam, Product info as of 2010.
14. David D. Boehr, Kari-ann Draker and Gerard D.Wright. Chapter 14. Aminoglycosides and aminocyclitols. In Roger Finch, David Greenwood, S.Norrby and Richard Whitlety,editors. Antibiotic and Chemotherapy. Anti-infectives agents and their use in therapy 2003. pp: 155-184.
15. Chapter 58 - Antimicrobial Therapy in the Critical Care Setting. Pp 659-674.
16. Roger L. Nation and Jian Li. Optimizing Use of Colistin and Polymyxin B in the Critically Ill. Semin Respir and Crit Care Med 2007; 28 (6): 604-614.
17. Koch-Weser J, Sidel VW, Federman EB, Kanarek P, Finer DC, Eaton AE. Adverse effects of sodium colistimethate: manifestations and specifi c reaction rates during 317 courses of therapy. Ann Intern Med1970; 72:857-68.
18. Brown JM, Dorman DC, Roy LP. Acute renal failure due to overdosage of colistin. Med J Aust 1970; 2: 923-924.
19. Ryan KJ, Schainuck LI, Hickman RO, Striker GE. Colistimethate toxicity: report of a fatal case in a previously healthy child. JAMA1969; 207: 2099-2101.
20. Charles-Edouard Luyt, Alain Combes, Ania Nieszkowska, Jean-Louis Trouillet and Jean Chastre. Aerosolized antibiotics to treat ventilator-associated pneumonia. Cu rrent Opinion in Infectious Diseases 2009; 22: 154-158.
21. Matthew E. Falagas and Sofi a K. Kasiakou. Colistin: The Revival of Polymyxins for the Management of Multidrug-Resistant Gram-Negative Bacterial Infections. Clin Infect Dis 2005; 40:1333-1341.
22. Coly-mycin M parenteral [package insert]. Bristol, TN: Monarch Pharmaceuticals, 2002.
23. Colomycin [package insert]. Bexley, UK: Forest Laboratories, UK Limited, 2002.
24. Argyris Michalopoulos and Matthew Falagas. Colistin: recent data on Pharmacodynamics properties and clinical effi cacy in critically ill patients. Ann Intensive Care 2011; 1(1): 30.
25. Francoise Van Bambeke, Yves Van Laethem, Patrice Courvalin and Paul M Tulkens. Glycopeptide Antibiotics from Conventional Molecules to new derivatives. Drugs 2004; 64(9): 913-936.
26. Tammy S. Lundstrom, Jack D. Sobel. Antibiotics for gram-positive bacterial infections: vancomycin, quinupristin-dalfopristin, linezolid, and daptomycin. Infect Dis Clin N Am 2004; 18: 651-668.
27. Vancocin CP, Product Info
28. Targocid, Product Info
29. Norrby R. Linezolid a review of the first oxazolidinone. Expert Opin Pharmacother 2001; 2(2):293-302.
30. Knut Ohlsen. Novel Antibiotics for the Treatment of Staphylococcus aureus. Expert Rev Clin Pharmacol 2009; 2(6): 661-672.
31. Zyvox, Prescribing Information as of 2010.
32. Philip I. Hair and Susan J. Keam. Daptomycin A Review of its Use in the Management of Complicated Skin and Soft-Tissue Infections and Staphylococcus aureus Bacteraemia. Drugs 2007; 67 (10): 1483-1512.
33. CUBICIN, Prescribing Information as of 2007.
34. David M. Livermore. Tigecycline: what is it, and where should it be used? J Antimicrob Chemother 2005; 56: 611-614.
35. Tygacil. Prescribing Information as of 2010.
36. John P. Manzella. Quinupristin-Dalfopristin: A New Antibiotic for Severe Gram-Positive Infections. Am Fam Physician 2001; 64(11):1863-1867.
37. Synercid, Prescribing Information as of November 2007.
38. April Barbour, Stephan Schmidt, Kenneth H. Rand, Hartmut Derendorf. Ceftobiprole: a novel cephalosporin with activity against Gram-positive and Gram-negative pathogens, including methicillin-resistant Staphylococcus aureus (MRSA). International Journal of Antimicrobial Agents 2009; 34:1-7.
39. Zhanel George G; Lam Ashley; Schweizer Frank; Thomson Kristjan; Walkty Andrew; Rubinstein Ethan; Gin Alfred S; Hoban Daryl J; Noreddin Ayman M; Karlowsky James A Ceftobiprole: a review of a broad-spectrum and anti-MRSA cephalosporin. Am J Clin Dermatology 2008; 9(4):245-54.
40. Shawn D Anderson, John G Gums. Ceftobiprole: An Extended-Spectrum Staphylococcus aureus Cephalosporin. Ann Pharmacother 2008; 42(6): 806-816.
41. George G Zhanel and Ayman M Noreddin. Pharmacokinetics and pharmacodynamics of the new fluoroquinolones: focus on respiratory infections. Curr Opin in Pharmacol 2001; 1(5):459-463.
42. Wael E. Shams and Martin E. Evans Guide to Selection of Fluoroquinolones in Patients with Lower Respiratory Tract Infections. Drugs 2005; 65 (7): 949-991.
43. Zhanel GG, Fontaine S, Adam H, Schurek K, Mayer M, Noreddin AM, Gin AS, Rubinstein E, Hoban DJ. A Review of New Fluoroquinolones: Focus on their Use in Respiratory Tract Infections. Treat Respir Med 2006; 5(6):437-465.
44. Shinzaburo MinamiI, Rikizou Hattori and Akira Matsuda. Pharmacological properties and expected clinical role of an injectable new quinolone antibiotic, pazufl oxacin mesilate. Nippon Yakuri Shi (Folia Pharmacol. Jpn) 2003; 122: 161-178.
45. Vincent T. Andriole. Chapter 29. Quinolones. In Roger Finch, David Greenwood, S.Norrby and Richard Whitlety, editors. Antibiotic and Chemotherapy. Anti-infectives agents and their use in therapy. Churchill Livingstone 2003; pp: 349-373.
46. Bryskier A. and Butzler P.J. Chapter 24. Macrolides. In Roger Finch, David Greenwood, S.Norrby and Richard Whitlety, editors. Antibiotic and Chemotherapy. Anti-infectives agents and their use in therapy. Churchill Livingstone 2003; pp: 310-325.
47. Willain .A. Petri, Jr. Chapter 43.Sulfonamides, Trimethoprim-Sulfamethoxazole, Quinolones and Agents for Urinary Tract Infections. In Laurence L. Brunton, John S.Lazo, Keith L. Parker, editors. Goodman and Gilman's. The Pharmacological Basics of Therapeutics. McGraw-Hill Medical Publishing Division 2006; 1111-1126.
48. Henry F. Chambers. Chapter 46. Protein Syntheis Inhibitors and Miscellaneous Antibacterial Agents. In Laurence L. Brunton, John S.Lazo, Keith L. Parker, editors. Goodman and Gilman's. The Pharmacological Basics of Therapeutics. McGraw-Hill Medical Publishing Division 2006; 1173-1202.
49. Wilcox.M.H. Chapter 18. Chloramphenicol and thiamphenicol. In Roger Finch, David Greenwood, S.Norrby and Richard Whitlety, editors. Antibiotic and Chemotherapy. Anti-infectives agents and their use in therapy. Churchill Livingstone 2003. pp: 279-283.
50. R.S.Satoskar, S.D. Bhandarkar, Nirmala N. Rege, editors. Pharmacology and Pharmacotherapeutics. 20th ed 2007. pp: 685-701.
51. Matthew E. Falagas, Konstantina P. Giannopoulou, George N. Kokolakis, 1 and Petros I. Rafailidis. Fosfomycin: Use beyond Urinary Tract and Gastrointestinal Infections. Clin Infect Dis 2008; 46:1069-1077.
52. Matthew E. Falagas, Antonia C. Kastoris, Drosos E. Karageorgopoulos, Petros I. Rafailidis. Fosfomycin for the treatment of infections caused by multidrug-resistant non-fermenting Gram-negative bacilli: a systematic review of microbiological, animal and clinical studies. Int J Antimicrob Agents 2009.
53. David R. Guay. An Update on the Role of Nitrofurans in the Management of Urinary Tract Infections. Drugs 2001; 61 (3): 353-364.
54. Richard Gleckman, Salvador Alvarez and Dennis W. Joubert. Drug therapy review: Nitrofurantoin. Am J Hosp Pharm 1979; 36:342-351.
55. Greenwood. D. Chapter 23 Lincosamides. In Roger Finch, David Greenwood, S. Norrby and Richard Whitlety, editors. Antibiotic and Chemotherapy. Anti-infectives agents and their use in therapy. Churchill Livingstone 2003. pp: 305-309.
56. Edwards I.D. Chapter 27 Nitroimidazoles. In Roger Finch, David Greenwood, S. Norrby and Richard Whitlety, editors. Antibiotic and Chemotherapy. Anti-infectives agents and their use in therapy. Churchill Livingstone 2003. pp: 335-344.
57. Chemotherapy 1974; 21: 113.
58. Thompson GR, Cadena J, Patterson TF. Overview of antifungal agents. Clin Chest Med 2009; 30: 203-215.
59. Treatment Guidelines from the Medical Letter. The Medical Letter 2008; 6:1.
60. The Lancet 2003; 362 (9390):1142-1151.
61. Drugs 2011; 71(1): 11-41.
62. Michael N Dudley, Jeff Loutit and David C Griffi th. Aerosol antibiotics: considerations in pharmacological and clinical evaluation. Curr Opin Biotechnol 2008, 19:637-643.
63. Amar Safdar, Samuel A Shelburne, Scott E Evans & Burton F Dickey. Inhaled therapeutics for prevention and treatment of pneumonia. Expert Opin Drug Saf (2009); 8(4):435-449.
64. Erik De Clercq. Antiviral drugs in current clinical use. Journal of Clinical Virology 2004; 30(2): 115-133.
65. Chapter 38 Antiviral Drugs in Lippincott's Illustrated Reviews: Pharmacology, 4th edition, editors Richard A. Harvey, Pamela C. Champe, pages 437-456.
66. Zovirax prescribing information as of 2003.
67. Chapter 49 Antiviral agents (non-retroviral). Editors Laurence Brunton, Donald Blumenthal, Keith Parker, Iain Buxton. Goodman and Gilman's Manual of Pharmacology and Therapeutics. McGraw-Hill Medical Publishing Division 2008, 812-836.
68. Michael N Dudley, Jeff Loutit and David C Griffi th. Aerosol antibiotics: considerations in pharmacological and clinical evaluation. Curr Opin Biotechnol 2008, 19:637-643.
69. Holly M. Mattoes, Joseph L. Kuti, George L. Drusano, and David P Nicolau. Optimizing Antimicrobial Pharmacodynamics: Dosage Strategies for Meropenem. Clin Ther 2004; 26(8):1187-1198.
70. Joan E. Kapusnik-Uner, Marjorie and Merle A. Sande. Chapter 58. Antimicrobial Therapy in the Critical Care Setting. In pp: 659-674.
2. Pramod M. Shah1 and Robin D. Isaacs. Ertapenem, the first of a new group of carbapenems. J Antimicrob Chemother 2003; 52: 538-542.
3. Joan E. Kapusnik-Uner, Marjorie and Merle A. Sande. Chapter 58 Antimicrobial Therapy in the Critical Care Setting. In pp: 659-674.
4. R.S.Satoskar, S.D. Bhandarkar, Nirmala N. Rege, editors. Pharmacology and Pharmacotherapeutics, 20th ed 2007. Pp: 648-668.
5. K.Bush. Chapter 16 Beta-Lactam Antibiotics: penicillins. In Roger Finch, David Greenwood, S.Norrby and Richard Whitlety, editors. Antibiotic and Chemotherapy. Anti-infectives agents and their use in therapy 2003; pp: 224-258.
6. William A Petri. Chapter 44 Penicillins, Cephalosporins and Other Beta-lactam Antibiotics. In Laurence L. Brunton, John S.Lazo, Keith L. Parker, editors. Goodman and Gilman's. The Pharmacological Basics of Therapeutics. McGraw-Hill Medical Publishing Division 2006; 1127-1154.
7. Nelson Lee, Kwok-Yung Yuen and Cyrus R. Kumana. Clinical Role of ?-Lactam/?-Lactamase Inhibitor Combinations. Drugs 2003; 63(14): 1511-1524.
8. Pramod M. Shah1 and Robin D. Isaacs. Ertapenem, the first of a new group of carbapenems. J Antimicrob Chemother 2003; 52: 538-542.
9. Katherine M. Knapp and B. Keith English. Carbapenems. Semin in Pedia Infect Dis 2001; 12(3): 175-185.
10. George G. Zhanel, Ryan Wiebe,Leanne Dilay, Kristjan Thomson, Ethan Rubinstein, Daryl J. Hoban, Ayman M. Noreddin and James A. Karlowsky. Comparative Review of the Carbapenems. Drugs 2007; 67(7): 1027-1052.
11. Kristen N Sch-Ryan Wiebe, lames A KarIowsky Ethan Rubinstein, Dary J Hoban and George Gzhanelt. Faropenem: review of a new oral penem. Expert Rev Infect Ther 2007; 5(2): 185-198.
12. Sutep Jaruratanasirikul, Somchai Sriwiriyajan and Jarurat Punyo. Comparison of the Pharmacodynamics of Meropenem in Patients with Ventilator-Associated Pneumonia following Administration by 3-Hour Infusion or Bolus Injection. Antimicrob Agents Chemother 2005; 49 (4):1337-1339.
13. Zienam, Product info as of 2010.
14. David D. Boehr, Kari-ann Draker and Gerard D.Wright. Chapter 14. Aminoglycosides and aminocyclitols. In Roger Finch, David Greenwood, S.Norrby and Richard Whitlety,editors. Antibiotic and Chemotherapy. Anti-infectives agents and their use in therapy 2003. pp: 155-184.
15. Chapter 58 - Antimicrobial Therapy in the Critical Care Setting. Pp 659-674.
16. Roger L. Nation and Jian Li. Optimizing Use of Colistin and Polymyxin B in the Critically Ill. Semin Respir and Crit Care Med 2007; 28 (6): 604-614.
17. Koch-Weser J, Sidel VW, Federman EB, Kanarek P, Finer DC, Eaton AE. Adverse effects of sodium colistimethate: manifestations and specifi c reaction rates during 317 courses of therapy. Ann Intern Med1970; 72:857-68.
18. Brown JM, Dorman DC, Roy LP. Acute renal failure due to overdosage of colistin. Med J Aust 1970; 2: 923-924.
19. Ryan KJ, Schainuck LI, Hickman RO, Striker GE. Colistimethate toxicity: report of a fatal case in a previously healthy child. JAMA1969; 207: 2099-2101.
20. Charles-Edouard Luyt, Alain Combes, Ania Nieszkowska, Jean-Louis Trouillet and Jean Chastre. Aerosolized antibiotics to treat ventilator-associated pneumonia. Cu rrent Opinion in Infectious Diseases 2009; 22: 154-158.
21. Matthew E. Falagas and Sofi a K. Kasiakou. Colistin: The Revival of Polymyxins for the Management of Multidrug-Resistant Gram-Negative Bacterial Infections. Clin Infect Dis 2005; 40:1333-1341.
22. Coly-mycin M parenteral [package insert]. Bristol, TN: Monarch Pharmaceuticals, 2002.
23. Colomycin [package insert]. Bexley, UK: Forest Laboratories, UK Limited, 2002.
24. Argyris Michalopoulos and Matthew Falagas. Colistin: recent data on Pharmacodynamics properties and clinical effi cacy in critically ill patients. Ann Intensive Care 2011; 1(1): 30.
25. Francoise Van Bambeke, Yves Van Laethem, Patrice Courvalin and Paul M Tulkens. Glycopeptide Antibiotics from Conventional Molecules to new derivatives. Drugs 2004; 64(9): 913-936.
26. Tammy S. Lundstrom, Jack D. Sobel. Antibiotics for gram-positive bacterial infections: vancomycin, quinupristin-dalfopristin, linezolid, and daptomycin. Infect Dis Clin N Am 2004; 18: 651-668.
27. Vancocin CP, Product Info
28. Targocid, Product Info
29. Norrby R. Linezolid a review of the first oxazolidinone. Expert Opin Pharmacother 2001; 2(2):293-302.
30. Knut Ohlsen. Novel Antibiotics for the Treatment of Staphylococcus aureus. Expert Rev Clin Pharmacol 2009; 2(6): 661-672.
31. Zyvox, Prescribing Information as of 2010.
32. Philip I. Hair and Susan J. Keam. Daptomycin A Review of its Use in the Management of Complicated Skin and Soft-Tissue Infections and Staphylococcus aureus Bacteraemia. Drugs 2007; 67 (10): 1483-1512.
33. CUBICIN, Prescribing Information as of 2007.
34. David M. Livermore. Tigecycline: what is it, and where should it be used? J Antimicrob Chemother 2005; 56: 611-614.
35. Tygacil. Prescribing Information as of 2010.
36. John P. Manzella. Quinupristin-Dalfopristin: A New Antibiotic for Severe Gram-Positive Infections. Am Fam Physician 2001; 64(11):1863-1867.
37. Synercid, Prescribing Information as of November 2007.
38. April Barbour, Stephan Schmidt, Kenneth H. Rand, Hartmut Derendorf. Ceftobiprole: a novel cephalosporin with activity against Gram-positive and Gram-negative pathogens, including methicillin-resistant Staphylococcus aureus (MRSA). International Journal of Antimicrobial Agents 2009; 34:1-7.
39. Zhanel George G; Lam Ashley; Schweizer Frank; Thomson Kristjan; Walkty Andrew; Rubinstein Ethan; Gin Alfred S; Hoban Daryl J; Noreddin Ayman M; Karlowsky James A Ceftobiprole: a review of a broad-spectrum and anti-MRSA cephalosporin. Am J Clin Dermatology 2008; 9(4):245-54.
40. Shawn D Anderson, John G Gums. Ceftobiprole: An Extended-Spectrum Staphylococcus aureus Cephalosporin. Ann Pharmacother 2008; 42(6): 806-816.
41. George G Zhanel and Ayman M Noreddin. Pharmacokinetics and pharmacodynamics of the new fluoroquinolones: focus on respiratory infections. Curr Opin in Pharmacol 2001; 1(5):459-463.
42. Wael E. Shams and Martin E. Evans Guide to Selection of Fluoroquinolones in Patients with Lower Respiratory Tract Infections. Drugs 2005; 65 (7): 949-991.
43. Zhanel GG, Fontaine S, Adam H, Schurek K, Mayer M, Noreddin AM, Gin AS, Rubinstein E, Hoban DJ. A Review of New Fluoroquinolones: Focus on their Use in Respiratory Tract Infections. Treat Respir Med 2006; 5(6):437-465.
44. Shinzaburo MinamiI, Rikizou Hattori and Akira Matsuda. Pharmacological properties and expected clinical role of an injectable new quinolone antibiotic, pazufl oxacin mesilate. Nippon Yakuri Shi (Folia Pharmacol. Jpn) 2003; 122: 161-178.
45. Vincent T. Andriole. Chapter 29. Quinolones. In Roger Finch, David Greenwood, S.Norrby and Richard Whitlety, editors. Antibiotic and Chemotherapy. Anti-infectives agents and their use in therapy. Churchill Livingstone 2003; pp: 349-373.
46. Bryskier A. and Butzler P.J. Chapter 24. Macrolides. In Roger Finch, David Greenwood, S.Norrby and Richard Whitlety, editors. Antibiotic and Chemotherapy. Anti-infectives agents and their use in therapy. Churchill Livingstone 2003; pp: 310-325.
47. Willain .A. Petri, Jr. Chapter 43.Sulfonamides, Trimethoprim-Sulfamethoxazole, Quinolones and Agents for Urinary Tract Infections. In Laurence L. Brunton, John S.Lazo, Keith L. Parker, editors. Goodman and Gilman's. The Pharmacological Basics of Therapeutics. McGraw-Hill Medical Publishing Division 2006; 1111-1126.
48. Henry F. Chambers. Chapter 46. Protein Syntheis Inhibitors and Miscellaneous Antibacterial Agents. In Laurence L. Brunton, John S.Lazo, Keith L. Parker, editors. Goodman and Gilman's. The Pharmacological Basics of Therapeutics. McGraw-Hill Medical Publishing Division 2006; 1173-1202.
49. Wilcox.M.H. Chapter 18. Chloramphenicol and thiamphenicol. In Roger Finch, David Greenwood, S.Norrby and Richard Whitlety, editors. Antibiotic and Chemotherapy. Anti-infectives agents and their use in therapy. Churchill Livingstone 2003. pp: 279-283.
50. R.S.Satoskar, S.D. Bhandarkar, Nirmala N. Rege, editors. Pharmacology and Pharmacotherapeutics. 20th ed 2007. pp: 685-701.
51. Matthew E. Falagas, Konstantina P. Giannopoulou, George N. Kokolakis, 1 and Petros I. Rafailidis. Fosfomycin: Use beyond Urinary Tract and Gastrointestinal Infections. Clin Infect Dis 2008; 46:1069-1077.
52. Matthew E. Falagas, Antonia C. Kastoris, Drosos E. Karageorgopoulos, Petros I. Rafailidis. Fosfomycin for the treatment of infections caused by multidrug-resistant non-fermenting Gram-negative bacilli: a systematic review of microbiological, animal and clinical studies. Int J Antimicrob Agents 2009.
53. David R. Guay. An Update on the Role of Nitrofurans in the Management of Urinary Tract Infections. Drugs 2001; 61 (3): 353-364.
54. Richard Gleckman, Salvador Alvarez and Dennis W. Joubert. Drug therapy review: Nitrofurantoin. Am J Hosp Pharm 1979; 36:342-351.
55. Greenwood. D. Chapter 23 Lincosamides. In Roger Finch, David Greenwood, S. Norrby and Richard Whitlety, editors. Antibiotic and Chemotherapy. Anti-infectives agents and their use in therapy. Churchill Livingstone 2003. pp: 305-309.
56. Edwards I.D. Chapter 27 Nitroimidazoles. In Roger Finch, David Greenwood, S. Norrby and Richard Whitlety, editors. Antibiotic and Chemotherapy. Anti-infectives agents and their use in therapy. Churchill Livingstone 2003. pp: 335-344.
57. Chemotherapy 1974; 21: 113.
58. Thompson GR, Cadena J, Patterson TF. Overview of antifungal agents. Clin Chest Med 2009; 30: 203-215.
59. Treatment Guidelines from the Medical Letter. The Medical Letter 2008; 6:1.
60. The Lancet 2003; 362 (9390):1142-1151.
61. Drugs 2011; 71(1): 11-41.
62. Michael N Dudley, Jeff Loutit and David C Griffi th. Aerosol antibiotics: considerations in pharmacological and clinical evaluation. Curr Opin Biotechnol 2008, 19:637-643.
63. Amar Safdar, Samuel A Shelburne, Scott E Evans & Burton F Dickey. Inhaled therapeutics for prevention and treatment of pneumonia. Expert Opin Drug Saf (2009); 8(4):435-449.
64. Erik De Clercq. Antiviral drugs in current clinical use. Journal of Clinical Virology 2004; 30(2): 115-133.
65. Chapter 38 Antiviral Drugs in Lippincott's Illustrated Reviews: Pharmacology, 4th edition, editors Richard A. Harvey, Pamela C. Champe, pages 437-456.
66. Zovirax prescribing information as of 2003.
67. Chapter 49 Antiviral agents (non-retroviral). Editors Laurence Brunton, Donald Blumenthal, Keith Parker, Iain Buxton. Goodman and Gilman's Manual of Pharmacology and Therapeutics. McGraw-Hill Medical Publishing Division 2008, 812-836.
68. Michael N Dudley, Jeff Loutit and David C Griffi th. Aerosol antibiotics: considerations in pharmacological and clinical evaluation. Curr Opin Biotechnol 2008, 19:637-643.
69. Holly M. Mattoes, Joseph L. Kuti, George L. Drusano, and David P Nicolau. Optimizing Antimicrobial Pharmacodynamics: Dosage Strategies for Meropenem. Clin Ther 2004; 26(8):1187-1198.
70. Joan E. Kapusnik-Uner, Marjorie and Merle A. Sande. Chapter 58. Antimicrobial Therapy in the Critical Care Setting. In pp: 659-674.

More From Area Of Interest
You may also like

5 Mar, 25

5 Mar, 25
Our Research
25 Jul, 24

25 Jul, 24
Our Research
23 Nov, 23
23 Nov, 23

2 Nov, 23
Our Research
5 Oct, 23

5 Oct, 23
Latest Items
6 Jun, 25
6 Jun, 25

6 Jun, 25


